 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2bh3 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Zn substituted E. coli Aminopeptidase P in complex with product | ||||||
 Components Components | XAA-PRO AMINOPEPTIDASE | ||||||
 Keywords Keywords | HYDROLASE / PROLINE-SPECIFIC PEPTIDASE / PRODUCT COMPLEX / METALLOENZYME / PITA-BREAD FOLD / DINUCLEAR HYDROLASE | ||||||
| Function / homology |  Function and homology information Function and homology informationXaa-Pro aminopeptidase / metalloexopeptidase activity / metalloaminopeptidase activity / aminopeptidase activity / manganese ion binding / protein homotetramerization / protein-containing complex / proteolysis / identical protein binding / cytosol Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.4 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.4 Å | ||||||
 Authors Authors | Graham, S.C. / Bond, C.S. / Freeman, H.C. / Guss, J.M. | ||||||
 Citation Citation | Journal: Biochemistry / Year: 2005 Title: Structural and Functional Implications of Metal Ion Selection in Aminopeptidase P, a Metalloprotease with a Dinuclear Metal Center. Authors: Graham, S.C. / Bond, C.S. / Freeman, H.C. / Guss, J.M. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2bh3.cif.gz | 104.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2bh3.ent.gz | 79.6 KB | Display | PDB format |
| PDBx/mmJSON format | 2bh3.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/bh/2bh3ftp://data.pdbj.org/pub/pdb/validation_reports/bh/2bh3 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1w2mC  1w7vC  1wbqC  1wl6C  1wl9C  1wlrC  2bhaC  2bhbC  2bhcC  2bhdC  2bn7C  1n51S S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |
























-Links
 PDBj
PDBj









- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| Unit cell |
| ||||||||
| Components on special symmetry positions |
|
-Components
-Protein , 1 types, 1 molecules A
| #1: Protein | Mass: 49744.062 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Details: DIPEPTIDE PRODUCT OF AMINOPEPTIDASE P (FORMED BY CLEVAGE OF XAA-PRO-LEU TRIPEPTIDE SUBSTRATE) Source: (gene. exp.) |
|---|
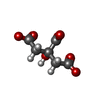
-Non-polymers , 6 types, 154 molecules 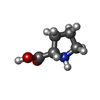




| #2: Chemical | ChemComp-PRO /  Type: L-peptide linking / Mass: 115.130 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C5H9NO2 Type: L-peptide linking / Mass: 115.130 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C5H9NO2 | ||||||
|---|---|---|---|---|---|---|---|
| #3: Chemical | ChemComp-LEU /  Type: L-peptide linking / Mass: 131.173 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H13NO2 Type: L-peptide linking / Mass: 131.173 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H13NO2 | ||||||
| #4: Chemical | ChemComp-ZN /  Mass: 65.409 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Zn Mass: 65.409 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Zn#5: Chemical | ChemComp-MG / |  Mass: 24.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mg Mass: 24.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mg#6: Chemical | ChemComp-FLC / |  Mass: 189.100 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H5O7 Mass: 189.100 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H5O7#7: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 146 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 5.32 Å3/Da / Density % sol: 76.7 % |
|---|---|
| Crystal grow | Temperature: 277 K / pH: 7 Details: AMINOPEPTIDASE P WAS DIALYSED AGAINST EGTA PRIOR TO CRYSTALLISATION. CRYSTALS WERE GROWN IN 26% MPD, 100 MM NACITRATE (PH 7.0) AND 200 MM MGACETATE AT 4C. CRYSTALS WERE SOAKED FOR 2 HOURS IN ...Details: AMINOPEPTIDASE P WAS DIALYSED AGAINST EGTA PRIOR TO CRYSTALLISATION. CRYSTALS WERE GROWN IN 26% MPD, 100 MM NACITRATE (PH 7.0) AND 200 MM MGACETATE AT 4C. CRYSTALS WERE SOAKED FOR 2 HOURS IN RESERVOIR SOLUTION SUPPLEMENTED WITH 1 MM ZNCL2 AND 10 MM PROLEU DIPEPTIDE PRIOR TO DATA COLLECTION. |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RU200H / Wavelength: 1.5418 |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE / Date: Dec 11, 2004 / Details: OSMIC MIRRORS |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 2.4→60 Å / Num. obs: 43466 / % possible obs: 99.2 % / Observed criterion σ(I): -3 / Redundancy: 6.1 % / Biso Wilson estimate: 55.02 Å2 / Rmerge(I) obs: 0.05 / Net I/σ(I): 25.2 |
| Reflection shell | Resolution: 2.4→2.49 Å / Redundancy: 5 % / Rmerge(I) obs: 0.52 / Mean I/σ(I) obs: 3.2 / % possible all: 98.5 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1N51 STRIPPED OF MULTIPLE CONFORMERS, SOLVENT ATOMS AND HETERO COMPOUNDS Resolution: 2.4→60.19 Å / Cor.coef. Fo:Fc: 0.964 / Cor.coef. Fo:Fc free: 0.954 / SU B: 8.434 / SU ML: 0.104 / TLS residual ADP flag: LIKELY RESIDUAL / Cross valid method: THROUGHOUT / ESU R: 0.159 / ESU R Free: 0.147 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS. SUFFICIENT DENSITY WAS NOT PRESENT FOR COMPLETE MODELLING OF RESIDUES A439 OR A440
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 47.17 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.4→60.19 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|