 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1okk | ||||||
|---|---|---|---|---|---|---|---|
| Title | HOMO-HETERODIMERIC COMPLEX OF THE SRP GTPASES | ||||||
 Components Components |
| ||||||
 Keywords Keywords | CELL CYCLE / SIGNAL RECOGNITION-COMPLEX / SRP / FFH / FTSY / GTPASE / MEMBRANE TARGETING / SIGNAL SEQUENCE RECOGNITION | ||||||
| Function / homology |  Function and homology information Function and homology informationsignal recognition particle / signal recognition particle binding / signal-recognition-particle GTPase / 7S RNA binding / SRP-dependent cotranslational protein targeting to membrane / GTPase activity / GTP binding / plasma membrane / cytoplasm Similarity search - Function | ||||||
| Biological species |   THERMUS AQUATICUS (bacteria) THERMUS AQUATICUS (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.05 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.05 Å | ||||||
 Authors Authors | Focia, P.J. / Freymann, D.M. | ||||||
 Citation Citation | Journal: Science / Year: 2004 Title: Heterodimeric Gtpase Core of the Srp Targeting Complex Authors: Focia, P.J. / Shepotinovskaya, I.V. / Seidler, J.A. / Freymann, D.M. #1: Journal: Acta Crystallogr.,Sect.D / Year: 2003 Title: Crystallization of the Gmppcp Complex of the Ng Domains of T. Aquaticus Ffh and Ftsy Authors: Shepotinovskaya, I.V. / Focia, P.J. / Freymann, D.M. #2: Journal: Biochim.Biophys.Acta / Year: 2002 Title: Conformational Change of the N-Domain on Formation of the Complex between the Gtpase Domains of Thermus Aquaticus Ffh and Ftsy Authors: Shepotinovskaya, I.V. / Freymann, D.M. #3: Journal: Structure / Year: 2001Title: The Conformation of Bound Gmppnp Suggests a Mechanism for Gating the Active Site of the Srp Gtpase Authors: Padmanabhan, S. / Freymann, D.M. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1okk.cif.gz | 141.3 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1okk.ent.gz | 107 KB | Display | PDB format |
| PDBx/mmJSON format | 1okk.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ok/1okkftp://data.pdbj.org/pub/pdb/validation_reports/ok/1okk | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1ng1S S: Starting model for refinement |
|---|---|
| Similar structure data |








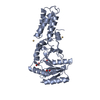










-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Details | FOR THE HETERO-ASSEMBLY DESCRIBED BY REMARK 350 |
-Components
-Protein , 2 types, 2 molecules AD
| #1: Protein | Mass: 32300.371 Da / Num. of mol.: 1 / Fragment: NG DOMAIN, RESIDUES 0-293 Source method: isolated from a genetically manipulated source Source: (gene. exp.) THERMUS AQUATICUS (bacteria) / Production host: |
|---|---|
| #2: Protein | Mass: 32972.230 Da / Num. of mol.: 1 / Fragment: NG DOMAIN, RESIDUES 2-304 Source method: isolated from a genetically manipulated source Source: (gene. exp.) THERMUS AQUATICUS (bacteria) / Production host: |
-Non-polymers , 6 types, 589 molecules 




| #3: Chemical |  Mass: 521.208 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C11H18N5O13P3 / Comment: GMP-PCP, energy-carrying molecule analogue*YM Mass: 521.208 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C11H18N5O13P3 / Comment: GMP-PCP, energy-carrying molecule analogue*YM#4: Chemical |  Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg#5: Chemical |  Mass: 130.145 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C5H10N2O2 Mass: 130.145 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C5H10N2O2#6: Chemical | ChemComp-SO4 /  Mass: 96.063 Da / Num. of mol.: 11 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 11 / Source method: obtained synthetically / Formula: SO4#7: Chemical | ChemComp-EDO /  Mass: 62.068 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C2H6O2 Mass: 62.068 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C2H6O2#8: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 568 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Details
| Sequence details | THE UNIPROT ENTRY O07347 HAS ANNOTATION |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.66 Å3/Da / Density % sol: 49.78 % | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 6.7 Details: 1.91 M AMMONIUM SULFATE, 0.1 M BISTRIS PH 6.7, 0.1 M NACL, 4% PEG 400. | ||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Method: other | ||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS  / Beamline: 5ID-B / Wavelength: 1 / Beamline: 5ID-B / Wavelength: 1 |
| Detector | Type: MARRESEARCH / Detector: CCD / Date: Feb 20, 2002 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 |
| Reflection | Resolution: 2.05→25 Å / Num. obs: 41332 / % possible obs: 99.9 % / Redundancy: 10.9 % / Rmerge(I) obs: 0.065 / Net I/σ(I): 33.4 |
| Reflection shell | Resolution: 2.05→2.1 Å / Redundancy: 10.4 % / Rmerge(I) obs: 0.158 / Mean I/σ(I) obs: 16.2 / % possible all: 100 |
| Reflection | *PLUS Highest resolution: 2.05 Å / Lowest resolution: 25 Å |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1NG1 Resolution: 2.05→24.73 Å / Cor.coef. Fo:Fc: 0.956 / Cor.coef. Fo:Fc free: 0.931 / SU B: 3.068 / SU ML: 0.086 / Cross valid method: THROUGHOUT / ESU R: 0.169 / ESU R Free: 0.146 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: THE N-TERMINAL THREE RESIDUES OF FFH (A1 - A3) ARE NOT VISIBLE IN THE ELECTRON DENSITY MAP. THE C- TERMINAL RESIDUE OF FFH (A 294) IS DISORDERED AND HAS BEEN OMITTED FROM THE MODEL. THE ...Details: THE N-TERMINAL THREE RESIDUES OF FFH (A1 - A3) ARE NOT VISIBLE IN THE ELECTRON DENSITY MAP. THE C- TERMINAL RESIDUE OF FFH (A 294) IS DISORDERED AND HAS BEEN OMITTED FROM THE MODEL. THE FIRST 20 AMINO ACIDS OF FTSY (CHAIN D) ARE PROTEOLYTICALLY CLEAVED AND ARE NOT ASSOCIATED WITH THE COMPLEX OR SEEN IN THE CRYSTAL STRUCTURE. FTSY (CHAIN D) IS PROTEOLYTICALLY CLEAVED AT RESIDUE 90, IN THE LINKER BETWEEN THE N AND G DOMAINS. RESIDUES D 79 - D 96 OF THE LINKER REGION ARE NOT VISIBLE IN THE ELECTRON DENSITY MAP. THE C-TERMINAL RESIDUE OF FTSY (D 304) IS NOT VISIBLE IN THE ELECTRON DENSITY MAP. ELECTRON DENSITY FOR FTSY LOOP D60 - D62 IS VERY POORLY DEFINED, AND THE CONFORMATION MAY BE INCORRECT. AN UNUSUAL PARTIAL RING OF ELECTRON DENSITY ENCIRCLES LYS D215 AND HAS BEEN MODELED AS TWO ETHYLENE GLYCOL MOLECULES, BUT SOME POSITIVE DIFFERENCE DENSITY REMAINS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: BABINET MODEL WITH MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 19.99 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.05→24.73 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|