Mass: 18.015 Da / Num. of mol.: 274 / Source method: isolated from a natural source / Formula: H2O
-
Details
Compound details
ESSENTIAL FOR EFFICIENT EXPORT OF EXTRA-CYTOPLASMIC PROTEINS. BINDS TO THE SIGNAL SEQUENCE OF ...ESSENTIAL FOR EFFICIENT EXPORT OF EXTRA-CYTOPLASMIC PROTEINS. BINDS TO THE SIGNAL SEQUENCE OF PROTEINS WHEN IT EMERGES FROM THE RIBOSOMES. TWO DOMAIN ARCHITECTURE: NG-DOMAIN BINDS GTP, M-DOMAIN BINDS RNA AND SIGNAL PEPTIDE OF THE PROTEIN TO BE TRANSPORTED. MEMBER OF THE SRP FAMILY OF GTP-BINDING PROTEINS
-
Experimental details
-
Experiment
Experiment
Method: X-RAY DIFFRACTION / Number of used crystals: 1
-
Sample preparation
Crystal
Density Matthews: 2.76 Å3/Da / Density % sol: 55.36 %
Resolution: 2.1→56.8 Å / Cor.coef. Fo:Fc: 0.941 / Cor.coef. Fo:Fc free: 0.913 / SU B: 4.928 / SU ML: 0.133 / Cross valid method: THROUGHOUT / ESU R: 0.22 / ESU R Free: 0.184 / Stereochemistry target values: MAXIMUM LIKELIHOOD / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS
Rfactor
Num. reflection
% reflection
Selection details
Rfree
0.237
2023
5.1 %
RANDOM
Rwork
0.195
-
-
-
obs
0.197
38000
97.1 %
-
Solvent computation
Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: BABINET MODEL WITH MASK
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information
 THERMUS AQUATICUS (bacteria)
THERMUS AQUATICUS (bacteria) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads






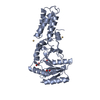






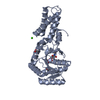



 PDBj
PDBj Assembly
Assembly







 Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl
Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl Mass: 46.025 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: CH2O2
Mass: 46.025 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: CH2O2 Type: RNA linking / Mass: 443.201 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H15N5O11P2 / Comment: GDP, energy-carrying molecule*YM
Type: RNA linking / Mass: 443.201 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H15N5O11P2 / Comment: GDP, energy-carrying molecule*YM Mass: 24.305 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Mg
Mass: 24.305 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Mg Sample preparation
Sample preparation / Beamline: 5ID-B / Wavelength: 0.96392
/ Beamline: 5ID-B / Wavelength: 0.96392  Processing
Processing