 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1jxu | ||||||
|---|---|---|---|---|---|---|---|
| Title | CRAMBIN MIXED SEQUENCE FORM AT 240 K. PROTEIN/WATER SUBSTATES | ||||||
 Components Components | Crambin | ||||||
 Keywords Keywords | PLANT PROTEIN / WATER / SUBSTATE / FUNCTION | ||||||
| Function / homology |  Function and homology information Function and homology information | ||||||
| Biological species |  Crambe hispanica subsp. abyssinica (Abyssinian crambe) Crambe hispanica subsp. abyssinica (Abyssinian crambe) | ||||||
| Method |  X-RAY DIFFRACTION / Resolution: 0.99 Å X-RAY DIFFRACTION / Resolution: 0.99 Å | ||||||
 Authors Authors | Teeter, M.M. / Yamano, A. / Stec, B. / Mohanty, U. | ||||||
 Citation Citation | Journal: Proc.Natl.Acad.Sci.USA / Year: 2001 Title: On the nature of a glassy state of matter in a hydrated protein: Relation to protein function. Authors: Teeter, M.M. / Yamano, A. / Stec, B. / Mohanty, U. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1jxu.cif.gz | 28 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1jxu.ent.gz | 18.5 KB | Display | PDB format |
| PDBx/mmJSON format | 1jxu.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/jx/1jxuftp://data.pdbj.org/pub/pdb/validation_reports/jx/1jxu | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1jxtC  1jxwC  1jxxC  1jxySC C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |








-Links
 PDBj
PDBj




- Assembly
Assembly
| Deposited unit | 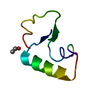
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Details | Biological assembly not known. |
-Components
| #1: Protein/peptide | Mass: 4728.410 Da / Num. of mol.: 1 / Source method: isolated from a natural source Source: (natural) Crambe hispanica subsp. abyssinica (Abyssinian crambe)Species: Crambe hispanica / Strain: subsp. abyssinica / References: UniProt: P01542 |
|---|---|
| #2: Chemical | ChemComp-EOH / 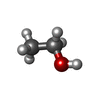  Mass: 46.068 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H6O Mass: 46.068 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H6O |
| Has protein modification | Y |
| Sequence details | sequence PRO/SER22:LEU/ILE25 SEQUENCE ISOFORMS ARE MODELLED AS ALTERNATE CONFORMERS IN COORDINATE ...sequence PRO/SER22:LEU/ILE25 SEQUENCE ISOFORMS ARE MODELLED AS ALTERNATE CONFORMERS |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 1.4 Å3/Da / Density % sol: 30 % Description: VALUES OF F/SIGF ARE GIVEN ABOVE RATHER THAN I/SIGI. |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, sitting drop / pH: 7 Details: ethanol, pH 7.00, VAPOR DIFFUSION, SITTING DROP, temperature 293K |
| Crystal | *PLUS |
| Crystal grow | *PLUS Method: unknown |
-Data collection
| Diffraction | Mean temperature: 240 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RU200 / Wavelength: 1.5418 |
| Detector | Type: RIGAKU AFC-5R / Detector: DIFFRACTOMETER / Date: Dec 25, 1990 / Details: COLLIMATOR |
| Radiation | Monochromator: MO / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 0.99→17.67 Å / Num. all: 7272 / Num. obs: 7272 / % possible obs: 92 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Redundancy: 1 % / Net I/σ(I): 17.2 |
| Reflection shell | Resolution: 0.99→1 Å / Redundancy: 1 % / Mean I/σ(I) obs: 4.4 / % possible all: 0.162 |
| Reflection | *PLUS % possible obs: 92 % / Rmerge(I) obs: 0.139 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Starting model: 1JXY. CRAMBIN MIXED ISOFORM AT 220 K. Resolution: 0.99→17.67 Å / Cross valid method: DIFFERENCE DENSITY PEAKS / σ(F): 2 / σ(I): 2 Details: OVERALL WEIGHT WAS 1/(SIGDEL) AND SIGDEL=8-10(SINTH/L-0.166667). WITH PROLSQ, NO RFREE WAS CALCULATED. ********* STEREOCHEMISTRY REMARKS: BECAUSE discretely disordered (MULTIPLE SUBSTATE) ...Details: OVERALL WEIGHT WAS 1/(SIGDEL) AND SIGDEL=8-10(SINTH/L-0.166667). WITH PROLSQ, NO RFREE WAS CALCULATED. ********* STEREOCHEMISTRY REMARKS: BECAUSE discretely disordered (MULTIPLE SUBSTATE) PROTEIN RESIDUES EACH HAVE OCCUPANCY LESS THAN ONE, THEY ARE DIFFICULT TO ACCURATELY REFINE. THE GEOMETRY CAN BE DISTORTED (IS POORLY DETERMINED).
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 5.5 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.06 Å / Luzzati d res low obs: 17.67 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 0.99→17.67 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: PROLSQ / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS σ(F): 2 / Rfactor obs: 0.09 / Rfactor Rwork: 0.09 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS Biso mean: 5.5 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
