 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1ab1 | ||||||
|---|---|---|---|---|---|---|---|
| Title | SI FORM CRAMBIN | ||||||
 Components Components | CRAMBIN (SER22/ILE25) | ||||||
 Keywords Keywords | PLANT SEED PROTEIN / THIONIN | ||||||
| Function / homology |  Function and homology information Function and homology information | ||||||
| Biological species |  Crambe hispanica subsp. abyssinica (Abyssinian crambe) Crambe hispanica subsp. abyssinica (Abyssinian crambe) | ||||||
| Method |  X-RAY DIFFRACTION / ALREADY SOLVED / Resolution: 0.89 Å X-RAY DIFFRACTION / ALREADY SOLVED / Resolution: 0.89 Å | ||||||
 Authors Authors | Teeter, M.M. / Yamano, A. | ||||||
 Citation Citation | Journal: J.Biol.Chem. / Year: 1997 Title: Crystal structure of Ser-22/Ile-25 form crambin confirms solvent, side chain substate correlations. Authors: Yamano, A. / Heo, N.H. / Teeter, M.M. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1ab1.cif.gz | 26.6 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1ab1.ent.gz | 17.7 KB | Display | PDB format |
| PDBx/mmJSON format | 1ab1.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ab/1ab1ftp://data.pdbj.org/pub/pdb/validation_reports/ab/1ab1 | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|




-Links
 PDBj
PDBj




- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein/peptide | Mass: 4728.410 Da / Num. of mol.: 1 / Source method: isolated from a natural source Source: (natural) Crambe hispanica subsp. abyssinica (Abyssinian crambe)Species: Crambe hispanica / Strain: subsp. abyssinica / References: UniProt: P01542 |
|---|---|
| #2: Chemical | ChemComp-EOH / 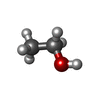  Mass: 46.068 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H6O Mass: 46.068 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H6O |
| Has protein modification | Y |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 1.77 Å3/Da / Density % sol: 30.37 % | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Method: vapor diffusion - hanging drop - microseeding / pH: 7 Details: HPLC PURIFIED SI-CRAMBIN PROTEIN WAS DISSOLVED AT 25 MG/ML IN 80% ETHANOL/WATER (V/V), AND EQUILIBRATED AGAINS 60% ETHANOL. AFTER 10 DAYS, THE RESERVOIR WAS LOWERED TO 50% AND MICROSEEDED BY ...Details: HPLC PURIFIED SI-CRAMBIN PROTEIN WAS DISSOLVED AT 25 MG/ML IN 80% ETHANOL/WATER (V/V), AND EQUILIBRATED AGAINS 60% ETHANOL. AFTER 10 DAYS, THE RESERVOIR WAS LOWERED TO 50% AND MICROSEEDED BY THE STREAK SEEDING METHOD (CAT WHISKER). AFTER LOWERING THE RESERVOIR TO 45%, GOOD CRYSTALS WERE FORMED., pH 7., vapor diffusion - hanging drop - microseeding | ||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 20 ℃ / Method: vapor diffusion / Details: Teeter, M.M., (1979) J. Mol. Biol., 127, 219. | ||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 150 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RUH2R / Wavelength: 1.5418 |
| Detector | Type: RIGAKU / Detector: IMAGE PLATE / Date: Oct 1, 1992 / Details: COLLIMATOR |
| Radiation | Monochromator: GRAPHITE(002) / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 0.89→17.67 Å / Num. obs: 23165 / % possible obs: 88.77 % / Observed criterion σ(I): 0 / Redundancy: 1 % / Rmerge(I) obs: 0.04 / Rsym value: 0.1 / Net I/σ(I): 8.3 |
| Reflection shell | Resolution: 0.89→0.897 Å / Redundancy: 0.1 % / Rmerge(I) obs: 0.1 / Mean I/σ(I) obs: 3.2 / Rsym value: 0.15 / % possible all: 74.96 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: ALREADY SOLVED / Resolution: 0.89→10 Å / Cross valid method: DIFFERENCE DENSITY PEAKS / σ(F): 2 Details: OVERALL WEIGHT WAS 1/(SIGDEL) AND SIGDEL=8-10(SINTH/L-0.166667) THIS STRUCTURE ILLUSTRATES THE CONFIRMATION OF THE ABILITY OF X-RAY DIFFRACTION TO DETECT SUBSTATES. HERE THE SUBSTATES WERE ...Details: OVERALL WEIGHT WAS 1/(SIGDEL) AND SIGDEL=8-10(SINTH/L-0.166667) THIS STRUCTURE ILLUSTRATES THE CONFIRMATION OF THE ABILITY OF X-RAY DIFFRACTION TO DETECT SUBSTATES. HERE THE SUBSTATES WERE PHYSICALLY SEPARATED (TWO SEQUENCE FORMS OF CRAMBIN) AND SEPARATE STRUCTURES DETERMINED. FURTHER SUBSTATES ARE EXTENDED FROM PROTEIN TO SURROUNDING SOLVENT.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 5.5 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.08 Å / Luzzati d res low obs: 17.6 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 0.89→10 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|
