 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1jxt | ||||||
|---|---|---|---|---|---|---|---|
| Title | CRAMBIN MIXED SEQUENCE FORM AT 160 K. PROTEIN/WATER SUBSTATES | ||||||
 Components Components | Crambin | ||||||
 Keywords Keywords | PLANT PROTEIN / WATER / SUBSTATE / FUNCTION | ||||||
| Function / homology |  Function and homology information Function and homology information | ||||||
| Biological species |  Crambe hispanica subsp. abyssinica (Abyssinian crambe) Crambe hispanica subsp. abyssinica (Abyssinian crambe) | ||||||
| Method |  X-RAY DIFFRACTION / AB INITIO PHASING / Resolution: 0.89 Å X-RAY DIFFRACTION / AB INITIO PHASING / Resolution: 0.89 Å | ||||||
 Authors Authors | Teeter, M.M. / Yamano, A. / Stec, B. / Mohanty, U. | ||||||
 Citation Citation | Journal: Proc.Natl.Acad.Sci.USA / Year: 2001 Title: On the nature of a glassy state of matter in a hydrated protein: Relation to protein function. Authors: Teeter, M.M. / Yamano, A. / Stec, B. / Mohanty, U. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1jxt.cif.gz | 30.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1jxt.ent.gz | 20.4 KB | Display | PDB format |
| PDBx/mmJSON format | 1jxt.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/jx/1jxtftp://data.pdbj.org/pub/pdb/validation_reports/jx/1jxt | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1jxuC  1jxwC  1jxxC  1jxyC  1cnrS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |






-Links
 PDBj
PDBj




- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Details | The biological assembly is not known. |
-Components
| #1: Protein/peptide | Mass: 4728.410 Da / Num. of mol.: 1 / Source method: isolated from a natural source Source: (natural) Crambe hispanica subsp. abyssinica (Abyssinian crambe)Species: Crambe hispanica / Strain: subsp. abyssinica / References: UniProt: P01542 |
|---|---|
| #2: Chemical | ChemComp-EOH / 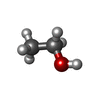  Mass: 46.068 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H6O Mass: 46.068 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H6O |
| Has protein modification | Y |
| Sequence details | sequence PRO/SER22:LEU/ILE25 SEQUENCE ISOFORMS ARE MODELLED AS ALTERNATE CONFORMERS IN COORDINATE ...sequence PRO/SER22:LEU/ILE25 SEQUENCE ISOFORMS ARE MODELLED AS ALTERNATE CONFORMERS |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 1.4 Å3/Da / Density % sol: 30 % Description: VALUES OF F/SIGF ARE GIVEN ABOVE RATHER THAN I/SIGI. |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, sitting drop / pH: 7 Details: ethanol, pH 7.00, VAPOR DIFFUSION, SITTING DROP, temperature 293K |
| Crystal | *PLUS |
| Crystal grow | *PLUS Method: unknown |
-Data collection
| Diffraction | Mean temperature: 160 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RU200 / Wavelength: 1.5418 |
| Detector | Type: RIGAKU AFC-5R / Detector: DIFFRACTOMETER / Date: Dec 7, 1990 / Details: COLLIMATOR |
| Radiation | Monochromator: MO / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 0.89→17.67 Å / Num. all: 26368 / Num. obs: 26368 / % possible obs: 96 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Redundancy: 1 % / Biso Wilson estimate: 3 Å2 / Net I/σ(I): 5.8 |
| Reflection shell | Resolution: 0.89→0.9 Å / Redundancy: 1 % / Mean I/σ(I) obs: 3.98 / Num. unique all: 704 / % possible all: 85 |
| Reflection | *PLUS % possible obs: 96 % / Rmerge(I) obs: 0.059 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: AB INITIO PHASING Starting model: 1CNR, PRO/LEU SEQUENCE FORM OF CRAMBIN. Resolution: 0.89→17.67 Å / Cross valid method: DIFFERENCE DENSITY PEAKS / σ(F): 2 Details: OVERALL WEIGHT WAS 1/(SIGDEL) AND SIGDEL=8-10(SINTH/L-0.166667). WITH PROLSQ, NO RFREE WAS CALCULATED. ********* STEREOCHEMISTRY REMARKS: BECAUSE discretely disordered (MULTIPLE SUBSTATE) ...Details: OVERALL WEIGHT WAS 1/(SIGDEL) AND SIGDEL=8-10(SINTH/L-0.166667). WITH PROLSQ, NO RFREE WAS CALCULATED. ********* STEREOCHEMISTRY REMARKS: BECAUSE discretely disordered (MULTIPLE SUBSTATE) PROTEIN RESIDUES EACH HAVE OCCUPANCY LESS THAN ONE, THEY ARE DIFFICULT TO ACCURATELY REFINE. THE GEOMETRY CAN BE DISTORTED (IS POORLY DETERMINED). IN THE CASE OF THR39, THE BACKBONE IS CLEARLY DISORDERED, WITH VERY SMALL DEVIATIONS BETWEEN SUBSTATES (~0.1 A), AND THAT WAS NOT INCLUDED IN THE MODEL. HENCE THEre may be LARGE ERRORS INVOLVING CA of thr39.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 3.58 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.06 Å / Luzzati d res low obs: 17.67 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 0.89→17.67 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: PROLSQ / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS σ(F): 2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
