 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1cbn: ATOMIC RESOLUTION (0.83 ANGSTROMS) CRYSTAL STRUCTURE OF THE HYDRO... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1cbn | ||||||
|---|---|---|---|---|---|---|---|
| Title | ATOMIC RESOLUTION (0.83 ANGSTROMS) CRYSTAL STRUCTURE OF THE HYDROPHOBIC PROTEIN CRAMBIN AT 130 K | ||||||
 Components Components | CRAMBIN | ||||||
 Keywords Keywords | PLANT SEED PROTEIN | ||||||
| Function / homology |  Function and homology information Function and homology information | ||||||
| Biological species |  Crambe hispanica subsp. abyssinica (Abyssinian crambe) Crambe hispanica subsp. abyssinica (Abyssinian crambe) | ||||||
| Method |  X-RAY DIFFRACTION / Resolution: 0.83 Å X-RAY DIFFRACTION / Resolution: 0.83 Å | ||||||
 Authors Authors | Teeter, M.M. / Roe, S.M. / Heo, N.H. | ||||||
 Citation Citation | Journal: J.Mol.Biol. / Year: 1993 Title: Atomic resolution (0.83 A) crystal structure of the hydrophobic protein crambin at 130 K. Authors: Teeter, M.M. / Roe, S.M. / Heo, N.H. #1: Journal: Acta Crystallogr.,Sect.B / Year: 1988Title: Cryocrystallography of Biological Macromolecules: A Generally Applicable Method Authors: Hope, H. #2: Journal: J.Am.Chem.Soc. / Year: 1986Title: An Empirical Examination of Potential Energy Minimization Using the Well-Determined Structure of the Protein Crambin Authors: Whitlow, M. / Teeter, M.M. #3: Journal: Ann.N.Y.Acad.Sci. / Year: 1986Title: Progress in the Water Structure of the Protein Crambin by X-Ray Diffraction at 140 K Authors: Teeter, M.M. / Hope, H. #4: Journal: Proc.Natl.Acad.Sci.USA / Year: 1984Title: Water Structure of a Hydrophobic Protein at Atomic Resolution. Pentagon Rings of Water Molecules in Crystals of Crambin Authors: Teeter, M.M. #5: Journal: Nature / Year: 1981Title: Structure of the Hydrophobic Protein Crambin Determined Directly from the Anomalous Scattering of Sulphur Authors: Hendrickson, W.A. / Teeter, M.M. #6: Journal: J.Mol.Biol. / Year: 1979Title: Highly Ordered Crystals of the Plant Seed Protein Crambin Authors: Teeter, M.M. / Hendrickson, W.A. | ||||||
| History |
| ||||||
| Remark 700 | SHEET IN SHEET *S1*, STRAND 3 IS QUESTIONABLY A STRAND. IN SHEET *S2*, STRAND 1 HAS NO HYDROGEN ...SHEET IN SHEET *S1*, STRAND 3 IS QUESTIONABLY A STRAND. IN SHEET *S2*, STRAND 1 HAS NO HYDROGEN BONDS. THE BACKBONE ATOMS ARE IN BETA CONFORMATION. |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1cbn.cif.gz | 28 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1cbn.ent.gz | 19.3 KB | Display | PDB format |
| PDBx/mmJSON format | 1cbn.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/cb/1cbnftp://data.pdbj.org/pub/pdb/validation_reports/cb/1cbn | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|




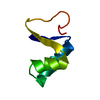

-Links
 PDBj
PDBj




- Assembly
Assembly
| Deposited unit | 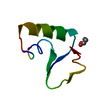
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein/peptide | Mass: 4728.410 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Crambe hispanica subsp. abyssinica (Abyssinian crambe)Species: Crambe hispanica / Strain: subsp. abyssinica / References: UniProt: P01542 |
|---|---|
| #2: Chemical | ChemComp-EOH / 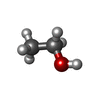  Mass: 46.068 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H6O Mass: 46.068 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H6O |
| Has protein modification | Y |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 1.78 Å3/Da / Density % sol: 30.9 % | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | *PLUS Method: vapor diffusion | ||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Radiation | Scattering type: x-ray |
|---|---|
| Radiation wavelength | Relative weight: 1 |
| Reflection | *PLUS Highest resolution: 0.826 Å / Lowest resolution: 10 Å / Num. all: 33221 / Num. obs: 26969 / Observed criterion σ(I): 2 / Rmerge(I) obs: 0.123 |
| Reflection shell | *PLUS Highest resolution: 0.83 Å / Lowest resolution: 0.85 Å / % possible obs: 91.5 % / Rmerge(I) obs: 0.215 |
- Processing
Processing
| Software | Name: PROLSQ / Classification: refinement | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Rfactor obs: 0.106 / Highest resolution: 0.83 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Highest resolution: 0.83 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: PROLSQ / Classification: refinement | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 0.83 Å / Lowest resolution: 10 Å / Rfactor obs: 0.106 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
