 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-4fc1: Ultra-high resolution neutron structure of crambin at room-temperature -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4fc1 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Ultra-high resolution neutron structure of crambin at room-temperature | ||||||
 Components Components | Crambin | ||||||
 Keywords Keywords | UNKNOWN FUNCTION / H/D exchange / neutron structure | ||||||
| Function / homology |  Function and homology information Function and homology information | ||||||
| Biological species |  Crambe hispanica subsp. abyssinica (Abyssinian crambe) Crambe hispanica subsp. abyssinica (Abyssinian crambe) | ||||||
| Method | NEUTRON DIFFRACTION / NUCLEAR REACTOR /  MOLECULAR REPLACEMENT / Resolution: 1.1 Å MOLECULAR REPLACEMENT / Resolution: 1.1 Å | ||||||
 Authors Authors | Kovalevsky, A.Y. / Chen, J.C.-H. | ||||||
 Citation Citation | Journal: Proc.Natl.Acad.Sci.USA / Year: 2012 Title: Direct observation of hydrogen atom dynamics and interactions by ultrahigh resolution neutron protein crystallography. Authors: Chen, J.C. / Hanson, B.L. / Fisher, S.Z. / Langan, P. / Kovalevsky, A.Y. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4fc1.cif.gz | 38.3 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4fc1.ent.gz | 27.2 KB | Display | PDB format |
| PDBx/mmJSON format | 4fc1.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/fc/4fc1ftp://data.pdbj.org/pub/pdb/validation_reports/fc/4fc1 | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|






-Links
 PDBj
PDBj


- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein/peptide | Mass: 4738.447 Da / Num. of mol.: 1 / Source method: isolated from a natural source Source: (natural) Crambe hispanica subsp. abyssinica (Abyssinian crambe)References: UniProt: P01542 |
|---|---|
| #2: Chemical | ChemComp-DOD / 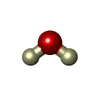  Mass: 18.015 Da / Num. of mol.: 42 / Source method: isolated from a natural source / Formula: D2O Mass: 18.015 Da / Num. of mol.: 42 / Source method: isolated from a natural source / Formula: D2O |
| Has protein modification | Y |
-Experimental details
-Experiment
| Experiment | Method: NEUTRON DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal grow | Temperature: 290 K Details: large crystals were obtained in 1mL crystallization drops containing 20-30mg/ml protein in 80% EtOH/H2O; the well solution was 60% EtOH/H2O; no mixing with well solution was done., VAPOR ...Details: large crystals were obtained in 1mL crystallization drops containing 20-30mg/ml protein in 80% EtOH/H2O; the well solution was 60% EtOH/H2O; no mixing with well solution was done., VAPOR DIFFUSION, SITTING DROP, temperature 290K |
|---|
-Data collection
| Diffraction | Mean temperature: 290 K | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: NUCLEAR REACTOR / Site: LANSCE  / Beamline: PCS / Wavelength: 0.7 - 6.0 / Beamline: PCS / Wavelength: 0.7 - 6.0 | |||||||||
| Detector | Type: HE3 POSITION SENSITIVE DETECTOR / Detector: AREA DETECTOR / Date: Oct 10, 2011 | |||||||||
| Radiation | Monochromator: NONE / Protocol: LAUE / Scattering type: neutron | |||||||||
| Radiation wavelength |
| |||||||||
| Reflection | Resolution: 1.1→15.18 Å / Num. obs: 11122 / % possible obs: 78.8 % / Observed criterion σ(I): 2 / Redundancy: 2.8 % / Rmerge(I) obs: 0.231 / Net I/σ(I): 5.5 | |||||||||
| Reflection shell | Resolution: 1.1→1.16 Å / Redundancy: 2 % / Rmerge(I) obs: 0.303 / Mean I/σ(I) obs: 1.8 / % possible all: 65.8 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT / Resolution: 1.1→10 Å / σ(F): 0 / Stereochemistry target values: ENGH & HUBER
| |||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.1→10 Å
| |||||||||||||||||||||||||||||||||
| Refine LS restraints |
|
