ジャーナル: EMBO J / 年: 2015 タイトル: X-ray and Cryo-EM structures reveal mutual conformational changes of Kinesin and GTP-state microtubules upon binding. 著者: Manatsu Morikawa / Hiroaki Yajima / Ryo Nitta / Shigeyuki Inoue / Toshihiko Ogura / Chikara Sato / Nobutaka Hirokawa / 要旨: The molecular motor kinesin moves along microtubules using energy from ATP hydrolysis in an initial step coupled with ADP release. In neurons, kinesin-1/KIF5C preferentially binds to the GTP-state ...The molecular motor kinesin moves along microtubules using energy from ATP hydrolysis in an initial step coupled with ADP release. In neurons, kinesin-1/KIF5C preferentially binds to the GTP-state microtubules over GDP-state microtubules to selectively enter an axon among many processes; however, because the atomic structure of nucleotide-free KIF5C is unavailable, its molecular mechanism remains unresolved. Here, the crystal structure of nucleotide-free KIF5C and the cryo-electron microscopic structure of nucleotide-free KIF5C complexed with the GTP-state microtubule are presented. The structures illustrate mutual conformational changes induced by interaction between the GTP-state microtubule and KIF5C. KIF5C acquires the 'rigor conformation', where mobile switches I and II are stabilized through L11 and the initial portion of the neck-linker, facilitating effective ADP release and the weak-to-strong transition of KIF5C microtubule affinity. Conformational changes to tubulin strengthen the longitudinal contacts of the GTP-state microtubule in a similar manner to GDP-taxol microtubules. These results and functional analyses provide the molecular mechanism of the preferential binding of KIF5C to GTP-state microtubules.
モード: BRIGHT FIELD / 倍率(公称値): 40000 X / 倍率(補正後): 40000 X / 最大 デフォーカス(公称値): 2600 nm / 最小 デフォーカス(公称値): 1200 nm / Cs: 3.3 mm
試料ホルダ
温度: 100 K / 傾斜角・最大: 0 ° / 傾斜角・最小: 0 °
撮影
電子線照射量: 10 e/Å2 / フィルム・検出器のモデル: KODAK SO-163 FILM
放射
プロトコル: SINGLE WAVELENGTH / 単色(M)・ラウエ(L): M / 散乱光タイプ: x-ray
放射波長
相対比: 1
-
解析
EMソフトウェア
ID
名称
バージョン
カテゴリ
1
MODELLER
モデルフィッティング
2
UCSF Chimera
モデルフィッティング
3
IMAGIC
5
3次元再構成
4
MATLAB
3次元再構成
CTF補正
詳細: Each filament
対称性
点対称性: C1 (非対称)
3次元再構成
手法: Single Particle / 解像度: 8.1 Å / 粒子像の数: 302000 / ピクセルサイズ(公称値): 2.5 Å / ピクセルサイズ(実測値): 2.5 Å / クラス平均像の数: 18 / 対称性のタイプ: POINT
原子モデル構築
プロトコル: RIGID BODY FIT / 空間: REAL Target criteria: Cross-correlation, Average map value, Atoms inside the contour 詳細: METHOD--Local refinement, Domain fitting REFINEMENT PROTOCOL--Rigid body refinement DETAILS--Initial local fitting was done using Chimera and for some loops Modeller was used.
 ムービー
ムービー コントローラー
コントローラー


 データを開く
データを開く
 基本情報
基本情報 要素
要素 キーワード
キーワード 機能・相同性情報
機能・相同性情報
 データ登録者
データ登録者 引用
引用

 構造の表示
構造の表示 ダウンロードとリンク
ダウンロードとリンク その他のダウンロード
その他のダウンロード












 PDBj
PDBj





 集合体
集合体



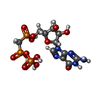

 分子量: 24.305 Da / 分子数: 2 / 由来タイプ: 合成 / 式: Mg
分子量: 24.305 Da / 分子数: 2 / 由来タイプ: 合成 / 式: Mg 分子量: 523.180 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C10H16N5O14P3 / コメント: GTP, エネルギー貯蔵分子*YM
分子量: 523.180 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C10H16N5O14P3 / コメント: GTP, エネルギー貯蔵分子*YM 分子量: 521.208 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C11H18N5O13P3
分子量: 521.208 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C11H18N5O13P3 分子量: 96.063 Da / 分子数: 1 / 由来タイプ: 合成 / 式: SO4
分子量: 96.063 Da / 分子数: 1 / 由来タイプ: 合成 / 式: SO4 試料調製
試料調製 電子顕微鏡撮影
電子顕微鏡撮影 FIELD EMISSION GUN / 加速電圧: 200 kV / 照射モード: FLOOD BEAM
FIELD EMISSION GUN / 加速電圧: 200 kV / 照射モード: FLOOD BEAM 解析
解析
