 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1lt8: Reduced Homo sapiens Betaine-Homocysteine S-Methyltransferase in ... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1lt8 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Reduced Homo sapiens Betaine-Homocysteine S-Methyltransferase in Complex with S-(delta-carboxybutyl)-L-Homocysteine | ||||||
 Components Components | BETAINE-HOMOCYSTEINE METHYLTRANSFERASE | ||||||
 Keywords Keywords | TRANSFERASE / homocysteine metabolism / homocysteinemia / zinc / thiol alkyl transfer | ||||||
| Function / homology |  Function and homology information Function and homology information: / betaine-homocysteine S-methyltransferase / amino-acid betaine metabolic process / betaine-homocysteine S-methyltransferase activity / L-methionine salvage / amino-acid betaine catabolic process / Sulfur amino acid metabolism / Choline catabolism / 'de novo' L-methionine biosynthetic process / protein methylation ...: / betaine-homocysteine S-methyltransferase / amino-acid betaine metabolic process / betaine-homocysteine S-methyltransferase activity / L-methionine salvage / amino-acid betaine catabolic process / Sulfur amino acid metabolism / Choline catabolism / 'de novo' L-methionine biosynthetic process / protein methylation / extracellular exosome / zinc ion binding / nucleus / cytosol Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.05 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.05 Å | ||||||
 Authors Authors | Evans, J.C. / Huddler, D.P. / Jiracek, J. / Castro, C. / Millian, N.S. / Garrow, T.A. / Ludwig, M.L. | ||||||
 Citation Citation | Journal: Structure / Year: 2002 Title: Betaine-homocysteine methyltransferase: zinc in a distorted barrel. Authors: Evans, J.C. / Huddler, D.P. / Jiracek, J. / Castro, C. / Millian, N.S. / Garrow, T.A. / Ludwig, M.L. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1lt8.cif.gz | 157.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1lt8.ent.gz | 122.2 KB | Display | PDB format |
| PDBx/mmJSON format | 1lt8.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/lt/1lt8ftp://data.pdbj.org/pub/pdb/validation_reports/lt/1lt8 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1lt7SC S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |








-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 44844.020 Da / Num. of mol.: 2 / Mutation: P2A, C104A, C131A, C186A, C201A, C256A Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Plasmid: pTYB4 / Species (production host): Escherichia coli / Production host:  References: UniProt: Q93088, betaine-homocysteine S-methyltransferase #2: Chemical |   Mass: 65.409 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Zn Mass: 65.409 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Zn#3: Chemical | 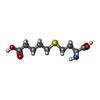  Mass: 235.301 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C9H17NO4S Mass: 235.301 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C9H17NO4S#4: Chemical |   Mass: 192.124 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C6H8O7 Mass: 192.124 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C6H8O7#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 431 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 431 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 2 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.07 Å3/Da / Density % sol: 40.72 % | ||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion, sitting drop / pH: 5.2 Details: PEG 200, sodium citrate, N,N-dimethylglycine, pH 5.20, VAPOR DIFFUSION, SITTING DROP, temperature 298K | ||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 22 ℃ | ||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS  / Beamline: 14-BM-C / Wavelength: 0.9 Å / Beamline: 14-BM-C / Wavelength: 0.9 Å |
| Detector | Type: ADSC QUANTUM 4 / Detector: CCD / Date: Nov 26, 2001 / Details: Bent conical Si-mirror (Rh coating) |
| Radiation | Monochromator: Bent Ge(111) monochromator / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9 Å / Relative weight: 1 |
| Reflection | Resolution: 2.05→100 Å / Num. all: 45962 / Num. obs: 45962 / % possible obs: 99.8 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Redundancy: 2.88 % / Biso Wilson estimate: 38.9 Å2 / Limit h max: 45 / Limit h min: -53 / Limit k max: 44 / Limit k min: -53 / Limit l max: 43 / Limit l min: 0 / Observed criterion F max: 2569593.7 / Observed criterion F min: 16.3 / Rmerge(I) obs: 0.057 / Rsym value: 0.057 / Net I/σ(I): 9.3 |
| Reflection shell | Resolution: 2.05→2.12 Å / Redundancy: 2.37 % / Rmerge(I) obs: 0.289 / Mean I/σ(I) obs: 2.46 / Num. unique all: 4569 / Rsym value: 0.289 / % possible all: 100 |
| Reflection | *PLUS Lowest resolution: 50 Å / Num. obs: 46046 / Redundancy: 2.9 % / Num. measured all: 131321 / Rmerge(I) obs: 0.057 |
| Reflection shell | *PLUS % possible obs: 100 % / Rmerge(I) obs: 0.289 / Mean I/σ(I) obs: 2.5 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 1lt7 Resolution: 2.05→10 Å / Rfactor Rfree error: 0.005 / Occupancy max: 1 / Occupancy min: 0.75 / Isotropic thermal model: Isotropic / Cross valid method: THROUGHOUT / σ(F): 0 / σ(I): 0 / Stereochemistry target values: Engh & Huber Details: Residues 326-331 from chain A in this model were generated from the model for chain B using non-crystallographic symmetry operators. Electron density in this region of the map is very poor.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: CNS bulk solvent model used / Bsol: 74.3159 Å2 / ksol: 0.41302 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 100.27 Å2 / Biso mean: 43.12 Å2 / Biso min: 11.96 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine Biso |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.05→10 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION / Total num. of bins used: 8
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: CNS / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Lowest resolution: 10 Å / % reflection Rfree: 5 % / Rfactor all: 0.236 / Rfactor Rfree: 0.255 / Rfactor Rwork: 0.225 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Rfactor Rfree: 0.337 / Rfactor Rwork: 0.296 |
