

IPR seminar : PDBj-BINDS joint workshop on January 30, 2020
IPR seminar : PDBj-BINDS joint workshop “Protein and Compound Structural Data for Drug Discovery and Life Science”
- Purpose
- Structural data for proteins and compounds are useful for drug design and life science to know mechanisms of molecular interactions. This BINDS workshop is organized by three units (protein function optimization unit, chemical seed and lead compounds exploratory unit, and in silico analysis unit) to know compound-protein interactions. The workshop provides PC-practices for compound-protein 3D data in PDB (Kawabata), comparisons of protein 3D structures (Rozewicki & Standley), and drug target database (Hijikata & Shirai). We also provide presentations for compound library and screening in Osaka university (Nunomura & Fujii), and in silico drug screening (Hirokawa).
- HOST
- Institute for Protein Research(IPR) and Basis for Supporting Innovative Drug Discovery and Life Science Research (BINDS)
- Date
- January 30, 2020, 10:30 - 18:00
- Place
- 1F Lecture hall in Institute for Protein Research (IPR), Osaka University
- Computer
- Bring your laptop PC with wireless LAN. Both Windows and Macintosh machines are available. We also lent a university laptop PC if you need to use it. We prepare about 15 PCs.
- Number of Participants
- About 40 persons
- Language
- Japanese and English
- Registration
- Please fill in application form.
- Deadline for registration
- January 29, 2020
- Contact us
- PDBj Contact Us
Program:Jan 30, 2020
- 10:30 - 10:35
- Opening(Genji Kurisu; IPR, Osaka Univ.)
- 10:35 – 12:00
-
“Searching and modeling compound-protein structure using HOMCOS”
Takeshi Kawabata (IPR, Osaka Univ.) - 12:00 - 13:00
- Lunch break
- 13:00 – 14:00
-
“Supports for Drug Discovery by the Drug Innovation Center in Osaka University”
Kazuto Nunomura, Shintaro Fujii (Drug Innovation Center, Grad. Sch. Pharm. Sci, Osaka Univ.) - 14:05 – 15:05
- "In silico Drug Discovery using Molecular Modeling and Simulation"
Takatsugu Hirokawa (molprof, AIST) - 15:25 – 16:25
-
"Application of structural alignment in immunology"
John Rozewicki, Daron Standley (BIKEN, Osaka Univ.) - 16:30 – 17:30
-
“Drug target discovery by Drug Target Excavator (DTX)”
Atsushi Hijigata, Tsuyosi Shirai (Nagahama-Bio. Univ.)
CiCLE workshop: sample evaluation for cryo-EM on January 6 or January 7, 2020
CiCLE workshop: sample evaluation for cryo-EM
Outline:For obtaining high-resolution structures by single particle cryo-EM suitable sample evaluation and sample preparation is essential. An important aspect of sample evaluation is the effective use of negative stain EM of the purified target protein. In this workshop lectures and hands-on practice workshops will provide an introduction to the use of negative stain EM observation of protein sample by conventional transmission electron microscopy (TEM) for the use in cryo-EM based structure determination.
- Date
- January 6 and 7, 2020 (Each participant attends one of the two days.)
- Place
- Institute for Protein Research (IPR), Osaka University
- Teachers
- Christoph Gerle, Genji Kurisu(IPR, Osaka University)
- Number of Participants
-
About five persons for each day. In total ten persons.
When more than 10 people apply, we would like to select participants, considering reasons of application and the order of application.
Persons who plan to use electron microscopy have priorities. - Where we will meet
- 9:00 AM at the 1F Lecture Hall in IPR
- Language
- Japanese and English
- Contact us
- PDBj Contact Us
Program:January 6 or January 7, 2020
(The programs of Jan 6 and Jan 7 are the same.)-
AM: Lectures [1F Lecture Hall]
- 9:15-9:30:
- General introduction(Kurisu)
- 9:30 - 10:50:
- Electron microcopy(Gerle)
- 11:00- 12:00:
- How to make and evaluate sample for EM(Gerle)
-
PM:Practices (Gerle, Misumi)
- 13:30 - 15:00:
- Making grids [3F Kurisu Lab]
- 15:10 - 17:00:
- Observing negative-staining samples using TEM H-7650
[1F TEM room in Research Center for the State-of-the-Art Functional Protein Analysis]
- Registration
- Please fill in application form.
- Deadline
- December 20, 2019
[wwPDB] Additional EM map validation now available through OneDep
The wwPDB validation reports provided in the OneDep system now have additional validation for electron microscopy (EM) maps to help users identify potential discrepancies in their data.
The updated wwPDB validation reports in the OneDep system now incorporate an extensive EM map validation process, integrating a range of established validation methods for EM data previously available on the EMDB pages. Initially, this additional EM validation is only provided to depositors in the OneDep system, however in future will be provided for entries throughout the PDB and EMDB archives.
The process includes an analysis of the fit of the PDB model to the EMDB map, represented at an amino acid level on the residue-property plots and globally by a visual overlay of the map and model (see below images). FSC curves are also included to compare reported and estimated resolution, where either half maps or FSC data was uploaded.
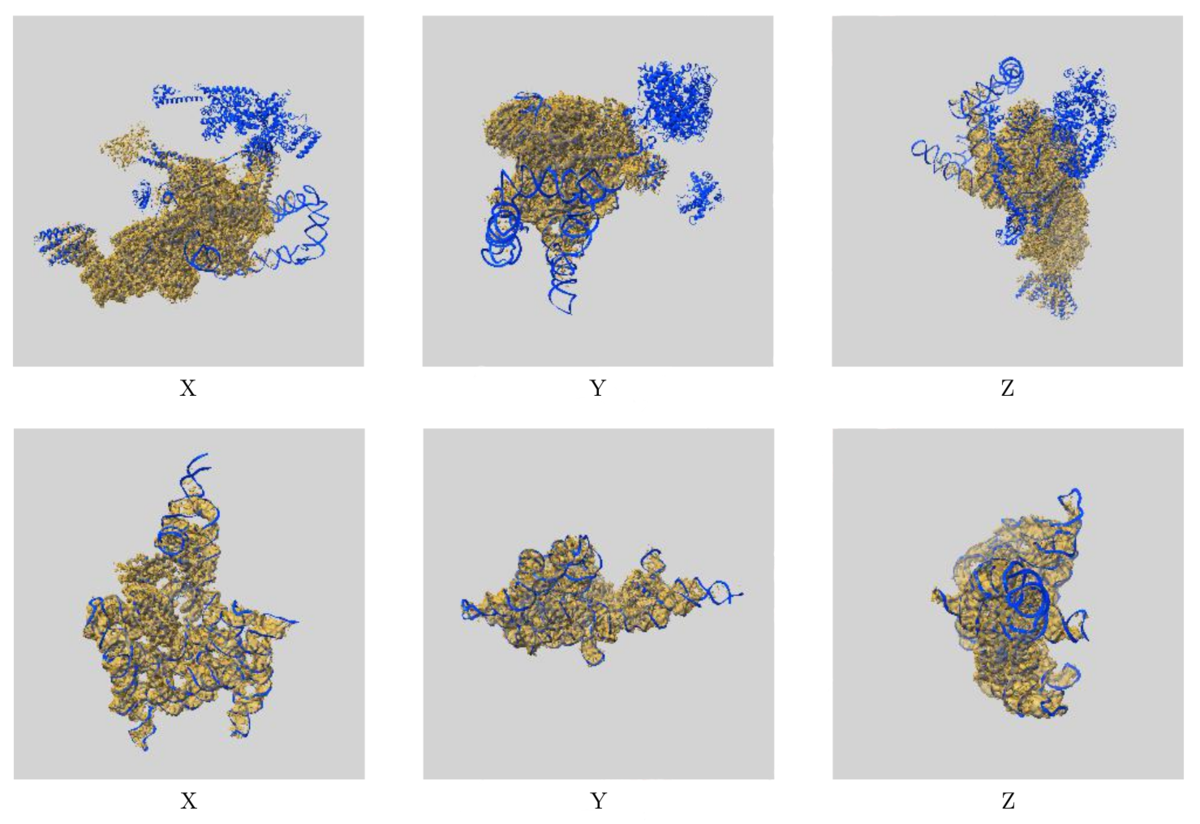 Images showing the visual EM map/model fit as displayed in the new validation reports.
The top images display three orthogonal views of the map and model for EMD-0360 and 6N7P and highlights where there are regions of the model
not covered by the EM map at the provided contour level, while the below images show the fit of EMD-9105 and 6ME0,
where the majority of the model fits well to the map at the provided contour level.
Images showing the visual EM map/model fit as displayed in the new validation reports.
The top images display three orthogonal views of the map and model for EMD-0360 and 6N7P and highlights where there are regions of the model
not covered by the EM map at the provided contour level, while the below images show the fit of EMD-9105 and 6ME0,
where the majority of the model fits well to the map at the provided contour level.
The additional EM validation also includes various graphics for visual inspection of the data. Included in the reports are images of orthogonal projections, central slices, mask visualisation, and more, allowing for inspection of details in the map and identification of artifacts. A statistical analysis of the EM map volume is also provided, including graphs of map density distribution, volume estimate by contour, and rotationally averaged power spectrum, providing more thorough analysis of the EM volume.
These changes should help both depositors and users to identify potential errors in EM data and give more clarity about potential limitations of the data in both the PDB and EMDB.
Additional information about validation reports for EM entries is available .
If you have any questions or queries about wwPDB validation, then please contact us at validation@mail.wwpdb.org.
[ wwPDB News ]
[wwPDB] Improved resolution of DOIs for PDB entries
The wwPDB partners are pleased to announce an improved mechanism for the resolution of digital object identifiers (DOIs) associated with released PDB entries. We have launched new wwPDB landing web-pages for each released PDB entry. These pages present basic information about the corresponding PDB structure, offer the model coordinate, experimental data and validation file downloads from the wwPDB FTP area, and, importantly, provide links to all the wwPDB partner websites that serve further advanced information about PDB structures. For an example, please navigate to the landing page for one of recently released entries in the PDB archive https://doi.org/10.2210/pdb6qw9/pdb. This development represents a significant improvement over the previous mechanism, where DOIs would resolve to a PDB-formatted file.
We encourage the scientific journals to make use of these pages and the DOIs issued for each PDB entry by linking to them from the online versions of papers where PDB entries are described or mentioned. We have also taken this opportunity to update the metadata that is associated with each PDB DOI, so that this information can be mined directly from the API offered by our DOI provider (CrossRef).
We would also like to draw the scientific community's attention to the new style of PDB accessions that are being gradually introduced, in preparation for when the supply of the familiar four character codes will be exhausted. The new accessions start with a prefix "PDB_" and contain further eight alphanumeric characters, with the last four characters identical to the familiar four-character codes.
[ wwPDB News ]
On 24th Sep, we’ll hold a luncheon seminar at the Annual Meeting of the Biophysical Society of Japan in Miyazaki
On 24th Sep, we will hold a luncheon seminar at the 57th Annual Meeting of the Biophysical Society of Japan in Miyazaki. Details are the following:
- Date
- 24th September 2019 (Tue) 11:30 - 12:20 (JST)
- Venue
- Seagaia Convention Center, Room G (IVORY) Hamayama, Yamazaki-cho, Miyazaki, 880-8545 Japan Access
- Fee
- Free (Separately, the registration fee is necessary)
- To participate
-
Registration at the annual meeting website is necessary. The application period is from 27th Aug 10:00 to 29th Aug 16:00 (JST). The participants are decided by drawing.
* A "Seminar Numbered Ticket" distribution desk will be set up from around 08:10 to 10:00 on the morning of the day, and lunch will be distributed to the first 10 out of the lottery members by the annual secretariat staff. If you would like to participate without lunch, please inquire about admission at the venue.
- Lecture Language
- English
- Lecture Title and Lecturer
-
-
"Recent activities of PDBj and wwPDB"
Genji Kurisu (Institute for Protein Research, Osaka University) -
"New tools for editing and annotating structural data"
Gert-Jan Bekker (Institute for Protein Research, Osaka University)
-
"Recent activities of PDBj and wwPDB"
- Additional Information
On 18th Aug (Sun), we exhibit a booth at the 29th Science Festa Osaka
We'll exhibit a booth at the 29th Science Festa Osaka, which is a scientific event for young people. The exhibition day of our booth is only on 8/18 (Sun).
We are going to exhibit the following menus:
- Explanation of the basic for proteins
- Experience the shape of proteins by stereo anaglyph
- Explanation of how to solve the shape of proteins
- Venue
- Herbis Hall (North Ward, Osaka, Official site of this hall, Google Map)
- Date
- 18th August 2019 (Sun) 10:00-17:00 (JST)
- Fee
- Free
[wwPDB] Improve your previously released coordinates AND keep your original PDB ID with OneDep
We are pleased to announce the availability of PDB versioning, allowing depositors to update their entries while retaining the same PDB accession code.
Depositors can now submit new coordinates for existing entries. Initially, this is limited to PDB entries that were submitted via the OneDep system, which was introduced in 2014. We plan to extend this functionality to entries deposited in the legacy systems (ADIT and Autodep) in future, and will announce a timeline for this in due course.
Requests should be initiated using the OneDep communication panel within the deposition session for the entry in question. Once submitted, the revised model will be processed by wwPDB biocurators and a new version released. Versioning of PDB entries will be limited to changes in the coordinate files, with no changes permitted to the deposited experimental data. PDB versioning will be limited to one replacement per PDB entry per year, and three entries per Principal Investigator per year.
The most recent version of the entry will be available in the main PDB archive FTP (ftp.pdbj.org [PDBj]/ ftp.wwpdb.org [wwPDB]). All major versions of a PDB structure will be retained in the versioned FTP archive (ftp-versioned.pdbj.org [PDBj]/ ftp-versioned.wwpdb.org [wwPDB]) - more information can be found on the wwPDB website. The structure of the versioned FTP archive has been built allowing for future extension of the PDB code format. PDB entry 1abc would therefore be found in the folder pdb_00001abc.
Changes made to entries during versioning are considered to be either “major” or “minor”. Updates to atomic coordinates, polymer sequence, or chemical description trigger a major version increment, while changes to any other categories are indicated as “minor”. Changes introduced are recorded in the PDBx/mmCIF audit categories.
If you have any further queries regarding the process of PDB versioning, please contact the wwPDB at deposit-help@mail.wwpdb.org.
[ wwPDB News ]
[wwPDB] From today: Mandatory PDBx/mmCIF format file submission for MX depositions
From today, July 1st 2019, submission of PDBx/mmCIF format files for crystallographic depositions to the PDB is mandatory.
PDBx/mmCIF is now the only format accepted for deposition of PDB structures resulting from macromolecular crystallography (MX), including X-ray, neutron, fiber, and electron diffraction methods via OneDep. The deposition of PDBx/mmCIF format files will improve the efficiency of the deposition process and enhance validation through capture of the more extensive experimental metadata supported by PDBx/mmCIF, compared to the legacy PDB format. PDB entries with 100,000 or more atoms, and those with multiple character chain IDs had no longer been supported by the legacy PDB format. In addition, by 2021, we anticipate the PDB Chemical Component Identifier will need to be extended beyond three characters, which will necessarily result in full retirement of files in the PDB Core Archive that utilize the legacy PDB format.
Refmac, Phenix.refine, and Buster programs can now output PDBx/mmCIF formatted files. For users of other structure determination/refinement software packages, the wwPDB provides stand-alone and web-based tools to convert legacy PDB format files into PDBx/mmCIF format: pdb_extract and MAXIT. More information on outputting and preparing PDBx/mmCIF format files for deposition can be found on the wwPDB website.
The PDBx/mmCIF Working Group has committed to the PDBx/mmCIF data model. PDBx/mmCIF is also supported by visualization software applications, including Jmol/JSMol, Chimera, OpenRasMol, CCP4MG, COOT, PyMOL, VMD, Molmil, LiteMol, Mol* and NGL. In addition, other data resources, such as the Protein Model Portal and SASBDB, have adopted and extended the PDBx/mmCIF framework for data representation.
For any further questions regarding deposition please contact the wwPDB on deposit-help@mail.wwpdb.org.
[ wwPDB News ]
[wwPDB] Improvements to visualization of ligand validation and electron density maps in the wwPDB validation report
Our recent update to the wwPDB validation reports provides much clearer validation information for ligands.
We now include 2-dimensional diagrams of ligands, highlighting geometric validation criteria and, for structures determined by crystallography, 3-dimensional views of electron density.
We also provide calculated electron density map coefficients which were used to generate the analysis in the validation reports.
Ligand Validation
We have collaborated with Global Phasing Ltd to integrate the ligand visualization from buster-report into the wwPDB validation report, as recommended by the wwPDB/CCDC/D3R Ligand Validation Workshop. The ligand visualization will be available for ligands that have been designated as "Ligand of Interest" by the depositor and ligands with a molecular weight greater than 250 Daltons that have outliers.
The following ligand instance of NAP was chosen intentionally as a representative of sub-optimal quality in both the ligand model and its agreement with the X-ray data.
Geometric analysis provided by CCDC Mogul will be highlighted on a 2D diagram of the ligand, as shown below.
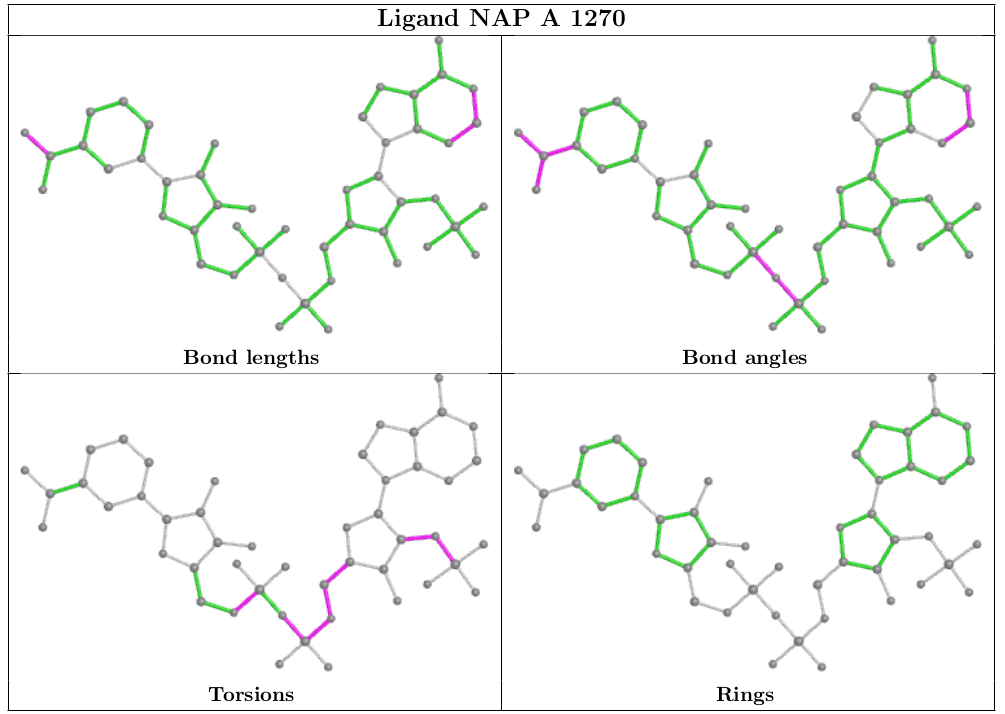
In addition to geometric validation for ligands, for X-ray diffraction PDB entries the wwPDB validation report also presents images displaying the ligand and the surrounding electron density map.
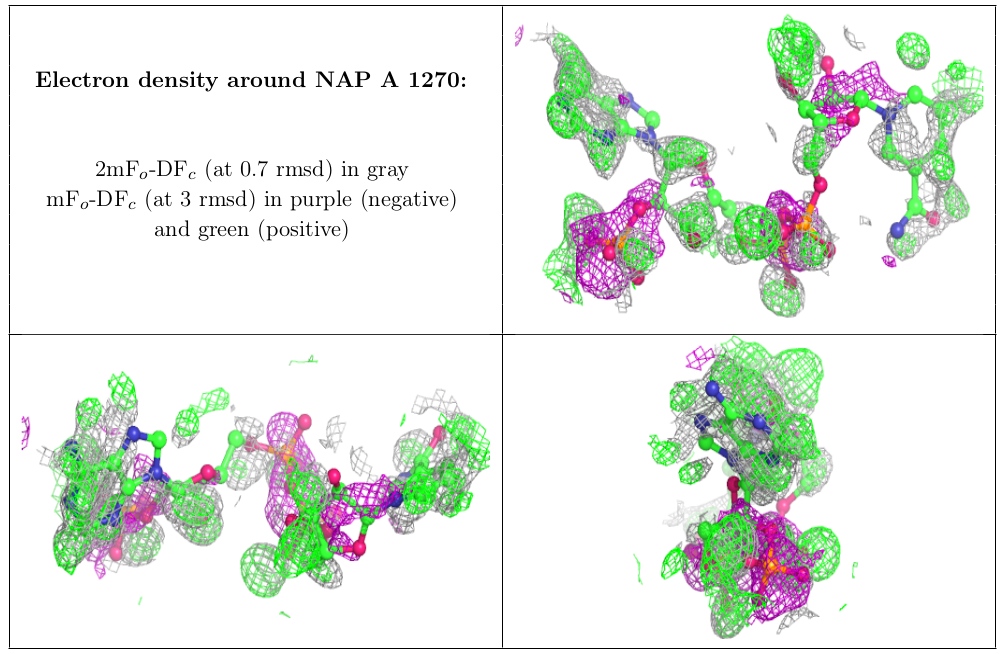
Electron Density Map Coefficient Files
We are now providing depositors with electron density map coefficient files (2mFo-DFc and mFo-DFc) from the wwPDB validation pipeline alongside the wwPDB validation report. The electron density map coefficients generated for wwPDB validation reports will be made available to end users in the PDB archive as new entries are released and for existing entries when validation reports for the PDB archive are recalculated.
We hope that these changes to the wwPDB validation pipeline will help depositors to interpret the validation information provided for PDB entries more easily. If you have any queries, please contact the wwPDB at deposit-help@mail.wwpdb.org.
[ wwPDB News ]
[wwPDB] A tribute to Prof. Michael G. Rossmann
All of us at wwPDB were deeply saddened to hear of the recent passing of Prof. Michael G. Rossmann and our condolences go out to his family and friends at this time. Michael contributed so much to the field of structural biology and was a great supporter of the wwPDB and our activities. Here we would like to share our own tribute to Michael and his contribution to the PDB archiving efforts.
Michael has had an esteemed structural biology career, with over 300 structures to his name in the PDB, spanning from 1977 through to 2019. He also helped to develop the molecular replacement technique, enabling phasing of data in X-ray crystallography, which has been used for structure determination of around 100,000 PDB entries to date. One of Michael's key discoveries is of the Rossmann fold, a structural motif commonly found in enzymes that bind to dinucleotide cofactors. It's importance is highlighted by the fact that around 20,000 structures in the PDB contain a Rossmann fold motif, another lasting legacy of his work within the PDB archive.
Michael also made huge contributions to the field of virology, with hundreds of his virus structures archived in the PDB. This includes the first structure determined of the human rhinovirus, otherwise known as the common cold. His work has significantly improved the knowledge of virus structure and function, supporting development of new therapeutics to treat and prevent viral infections.
Michael was a member of the wwPDB advisory committee for four years, serving on the board from 2009 to 2012, and was also involved in advisory committees for RCSB PDB and EMDataBank. During this time, he provided much advice and guidance to the wwPDB, helping to shape the archiving activities during this time and into the future. Helen Berman, former director of the RCSB Protein Data Bank, highlights how Michael "was an active supporter of the Protein Data Bank from its inception in the early 1970s", while Haruki Nakamura, former head of Protein Data Bank Japan (PDBj), speaks of "always being impressed by his thoughtful comments about the activities of the wwPDB." Haruki adds: "Michael understood the importance of archiving raw experimental data and he had promoted 'data science' for many years."
Gerard Kleywegt, former head of Protein Data Bank in Europe (PDBe), pays tribute to Michael as a person, describing him as "a dynamo, with an enormous zest for life and science who got along with almost everybody." Gerard also emphasises how, even in recent months, "he was still collecting data himself" and recalls Michael's dismay at a grant funding rejection and how "he simply could not understand how that was possible as it was obviously solid science."
We are indebted to Michael for his help and support to the wwPDB and to the structural biology community as a whole. As Helen Berman puts it: "Michael was truly unique in his contributions to structural biology and we will be forever grateful for all he did."

wwPDB AC in 2012
[ wwPDB News ]



