A new portal site that connects Japanese gene mutation information published in PDBj with protein sequences and structures (e.g., UniProt ID: P20813)
The Protein Data Bank Japan (PDBj, head Prof. Genji Kurisu)*1, operated by the Institute for Protein Research at Osaka University, and the Tohoku Medical Megabank Organization (ToMMo, head Prof. Masayuki Yamamoto)*2 at Tohoku University have jointly developed and launched a new portal as part of PDBj’s services (https://pdbj.org/uniprot/).
The portal not only provides links to genetic variants from the Japanese Multi Omics Reference Panel (jMorp, head Prof. Kengo Kinoshita)*3, developed by ToMMo, but also includes a tool that facilitates easy visualization of these variants on the 3D structure of proteins. Mapping genetic variants to the 3D structure of proteins is typically a complex task, but this new portal significantly simplifies the process.
The portal compiles the 3D structures of proteins registered in the PDB based on their UniProt IDs. It ranks these structures using a scoring system that takes into account factors such as resolution and the coverage of UniProt amino acid sequences for which 3D structural data is available. This enables users to efficiently select and analyze the relevant structures.
Protein Data Bank Japan (PDBj) was founded in 2000 at the Institute for Protein Research, Osaka University, as an organization responsible for the activities of the Joint Usage and Research Center. For 20 years, it has collected, edited, and registered information on the three-dimensional structures of proteins and nucleic acids from Asia and the Middle East, and in cooperation with the US RCSB, BMRB, and the European PDBe, has been disseminating all data from the University of Osaka to the world as a single internationally unified protein structure database (PDB).
*2 Tohoku Medical Megabank Organization:ToMMo head Prof. Masayuki Yamamoto
Founded in 2012 with the aim of recovery from the Great East Japan Earthquake and the realization of personalized prevention and medical care, this organization is promoting the Tohoku Medical Megabank Project together with the Iwate Tohoku Medical Megabank Organization at Iwate Medical University. The Tohoku Medical Megabank Project has been developing a biobank that has collected samples and information by conducting a resident cohort survey and a three-generation cohort survey on 150,000 people since 2013. In addition, the organization is developing a system and building a database so that the samples and information from the biobank can be utilized by both industry and academia. Since 2015, the Japan Agency for Medical Research and Development (AMED) has been the research support organization for this project.
*3 Japanese Multi Omics Reference Panel:jMorp head Prof. Kengo Kinoshita
The Japanese Multi-Omics Reference Panel (jMorp) is a website operated by the Tohoku Medical Megabank Organization (ToMMo) at Tohoku University that analyzes genome and omics data from participants in the cohort study of the Tohoku Medical Megabank Project and publishes the resulting statistical data. jMorp was launched in July 2015 as a database to publish the results of metabolomic and proteomic data analysis and has since been updated annually to include genome, transcriptome, metagenomics, and other data.
Created: 2024-11-01(last edited: 2 days ago)2024-11-01
294 new PDB entries have been released on 2024-10-30.
294 new PDB entries have been released on 2024-10-30.
226707 entries are now available in total.
We will hold a luncheon seminar at APBJC 2024 (1st Asia & Pacific Bioinformatics Joint Conference ~JSBi, GIW, InCoB, APBC, and ISCB-Asia~) on Thursday, October 24, 2024. We look forward to seeing you there.
Date
24th October 2024 (Thu) 12:30 - 13:30 (JST = UTC+9)
The PDBj (PDB Japan, https://pdbj.org/) is a core member of the worldwide Protein Data Bank (wwPDB, https://wwpdb.org/) and processes the deposited structural data from researchers in Asian and Middle-east regions. In order to promote the recent "Data Science", the wwPDB is introducing several new policies: (i) Collection of ORCID (Open Researcher and Contributor ID: http://orcid.org/) for contact authors and login using ORCID has started already, (ii) Visualization of ligand validation and electron density maps in the wwPDB validation report was improved, (iii) The NMR exchange (NEF) and NMR-STAR formats provide a standardized approach for representing commonly used NMR restraints. Using these restraint formats, a standardized validation system for assessing structural models of biopolymers against restraints has been developed and implemented in the wwPDB OneDep data harvesting system. These issues will be introduced at the Seminar.
The Royal Swedish Academy of Sciences has announced that they will award David Baker, Demis Hassabis,
and John Jumper the Nobel Prize in Chemistry 2024
for computational protein design and protein structure prediction.
More than 60,000 PDB depositors have submitted experimental data that are carefully reviewed, validated, and biocurated by the wwPDB team.
wwPDB partners adhere to the FAIR principles of Findability, Accessibility, Interoperability, and Reusability,
and ensure that all archival data can be accessed at no charge and with no limitations on usage under the most permissive
Creative Commons CC0 1.0 Universal License.
These PDB data provided training sets for developing protein design and prediction methods recognized by this award.
The models generated by AlphaFold and RoseTTAfold produce remarkably accurate 3D structures based on a given sequence.
In turn, these models are driving new PDB depositions, as they can be used for molecular replacement in MX
or combined with 3DEM density maps to reveal atomic details at a level and quality often unattainable by prediction
or experimentation alone.
Also notable is the impact of PDBx/mmCIF, which has become the data standard
for structural biology. PDBx/mmCIF provides an extensible data representation that is used for deposition
and archiving of experimentally determined 3D structures of biological macromolecules by the wwPDB.
Extensions of PDBx/mmCIF (ModelCIF)
are similarly used for computed structure models by ModelArchive,
for models computed with the AlphaFold 2.0 deep learning software suite, and the
AlphaFold Database.
The wwPDB congratulates these researchers and their teams for their revolutionary achievements that have changed the field.
Preprint publications will trigger wwPDB data archive release
The wwPDB releases entries under the following circumstances:
upon author’s request
upon publication
at the end of the hold period defined during submission (up to 1 year) if no publication is available by that time
Contributions to public preprint archives that reference PDB, EMDB, or BMRB entry IDs are considered publications
by the wwPDB and will therefore trigger release.
For example, a PDB structure on hold for publication (status HPUB/HOLD) will be scheduled for release
if the wwPDB finds a bioRxiv preprint with matching authors, title, and an entry ID code.
Publication dates and citation details are obtained through a combination of direct communications
from authors, journals, and members of the scientific community
(communicated via OneDep or deposit-help@mail.wwpdb.org)
and PubMed searches (automated comparison of title and author lists included with the deposition
and manual review for PDB, EMDB, and BMRB IDs).
Any concerns regarding this policy should be directed to the wwPDB leadership via
deposit-help@mail.wwpdb.org.
The PDB archive is now including annotation of protein chemical modifications (PCMs)
and post-translational modifications (PTMs) in a standardized way.
A new category called
pdbx_chem_comp_pcm, stating the PCM/PTM
type
and category,
as well as on which positions in the amino acid and in the polypeptide it is expected to be observed.
If this PCM is also a known PTM, it will have the Uniprot PTM accession ID.
A new category called
pdbx_modification_feature,
providing an instance-level annotation of all observed PCMs/PTMs within the entry, as well as their type and category.
Additionally to providing this new annotation, any protein modifications that are inconsistently handled within
PDB entries are amended, to ensure that a given modification is consistently handled in the PDB archive.
This includes a major clean-up of incorrect link records (struct_conn).
All entries containing protein modifications are being re-released gradually from October 2024, throughout Spring 2025.
This standardization ensures that there is a single approach to handling each protein modification that occurs
within the PDB archive, allowing better findability.
The protein chemical modifications (PCMs) and post translational modifications (PTMs) remediation project is
a wwPDB collaborative project carried out principally by PDBe
at EMBL-EBI, and is funded by BBSRC grant number BB/V018779/1.
Integrative structures are available at wwPDB.org and the PDB archive
Structures determined by integrative and hybrid structure determination methods (IHM) are now available
at wwPDB DOI landing pages for both released and on-hold entries, along with >225,000 experimental structures in the PDB archive.
These pages present basic information about the corresponding IHM structure,
offer download of model coordinates and validation files from the PDB archive
(https://files.wwpdb.org/pub/pdb_ihm/), and provide a link to the
PDB-Dev resource that currently serves more detailed information about IHM structures,
including the newly available links to PDB DOIs.
For an example, visit the DOI landing page for a recently-released IHM entry in the PDB archive via PDB DOI:
https://doi.org/10.2210/pdb9a8n/pdb.
PDB DOIs issued for each IHM or PDB entry are linked from the online versions of papers where PDB IDs are mentioned.
Users can distinguish IHM structures from PDB experimental structures on the DOI landing page where IHM structures have “integrative” as structure determination method displayed.
The FTP protocol for file downloads has been losing popularity over the years in favor of HTTP/S.
There are many advantages of HTTP/S including speed, statelessness, security (HTTPS), and better support.
Importantly during the past 2-3 years the main web browsers (Chrome and Firefox) have dropped support
for the FTP protocol, which has effectively discontinued the FTP protocol for non-technical users.
Given that the majority of file download activity on the internet has moved to HTTP/S,
wwPDB plans to deprecate FTP download protocol on November 1st 2024
(see previous announcement).
Support for the RSYNC protocol, which offers additional functionality, will continue to be maintained.
[wwPDB] Paper Published on CryoEM Archiving and Validation Recommendations
A workshop was held at EMBL-EBI (Hinxton, UK) in January 2020 to discuss data requirements for deposition and validation
of cryoEM structures, with a focus on single-particle analysis and setting community recommendations.
The outcomes of this meeting have now been published in this manuscript which highlights the recent achievements
made by the wwPDB in the space of 3DEM validation and the community recommendations going forward.
Some of these recommendations have already been implemented, such as a three-tiered strategy powered by
the Validation Analysis (VA) pipeline for the dissemination of validation information and ensuring the
that VA can be run by external applications.
Community recommendations on cryoEM data archiving and validation
Gerard J. Kleywegt, Paul D. Adams, Sarah J. Butcher, Cathy Lawson, Alexis Rohou, Peter B. Rosenthal,
Sriram Subramaniam, Maya Topf, Sanja Abbott, Philip R. Baldwin, John M. Berrisford, Gérard Bricogne,
Preeti Choudhary, Tristan I. Croll, Radostin Danev, Sai J. Ganesan, Timothy Grant, Aleksandras Gutmanas,
Richard Henderson, J. Bernard Heymann, Juha T. Huiskonen, Andrei Istrate, Takayuki Kato, Gabriel C. Lander,
Shee-Mei Lok, Steven J. Ludtke, Garib N. Murshudov, Ryan Pye, Grigore D. Pintilie, Jane S. Richardson,
Carsten Sachse, Osman Salih, Sjors H.W. Scheres, Gunnar F. Schroeder, Carlos Oscar S. Sorzano, Scott M. Stagg,
Zhe Wang, Rangana Warshamanage, John D. Westbrook, Martyn D. Winn, Jasmine Y. Young, Stephen K. Burley, Jeffrey C. Hoch,
Genji Kurisu, Kyle Morris, Ardan Patwardhan, Sameer Velankar (2024) IUCrJ 11: 140–151
https://doi.org/10.1107/S2052252524001246
[wwPDB] Paper Published on NMR Restraint Validation
We are pleased to announce the publication of this manuscript, addressing the challenge of validation of
experimental biomolecular NMR structures against restraint data.
The NMR exchange (NEF) and NMR-STAR formats provide a standardized approach for representing commonly used NMR restraints.
Using these restraint formats, a standardized validation system for assessing structural models of biopolymers
against restraints has been developed and implemented in the wwPDB OneDep data harvesting system.
The resulting wwPDB Restraint Violation Report provides a model vs data assessment of biomolecule structures
determined using distance and dihedral restraints, with extensions to other restraint types currently being implemented.
These tools are useful for assessing NMR models, as well as for assessing biomolecular structure predictions
based on distance restraints.
We present the rationale for model-vs-data restraint validation by the wwPDB, together with summary of
validation tools and reports for NMR distance and dihedral restraints that have been developed,
as implemented in the wwPDB validation pipeline and recommended by the wwPDB NMR-VTF committee.
Restraint Validation of Biomolecular Structures Determined by NMR in the Protein Data Bank
Kumaran Baskaran, Eliza Ploskon, Roberto Tejero, Masashi Yokochi, Deborah Harrus, Yuhe Liang, Ezra Peisach, Irina Persikova,
Theresa A Ramelot, Monica Sekharan, James Tolchard, John D Westbrook, Benjamin Bardiaux, Charles Schwieters, Ardan Patwardhan,
Sameer Velankar, Stephen K Burley, Genji Kurisu, Jeffrey C Hoch, Gaetano T Montelione, Geerten W Vuister, Jasmine Y Young
(2024) Structure 32, 1–14: doi: 10.1016/j.str.2024.02.011
The wwPDB plans to further enhance validation report by providing model-vs-data quality assessment for other kinds of restraints
based on community recommendation and improve data representation on structures with multiple conformation states.
A new paper describes how the recently-announced NextGen Archive
provides centralized access to integrated annotations and enriched structural information for PDB data:
NextGen Archive: Centralising Access to Integrated Annotations and Enriched Structural Information
by the Worldwide Protein Data Bank
Preeti Choudhary, Zukang Feng, John Berrisford, Henry Chao, Yasuyo Ikegawa, Ezra Peisach, Dennis W. Piehl, James Smith,
Ahsan Tanweer, Mihaly Varadi, John D. Westbrook, Jasmine Y. Young, Ardan Patwardhan, Kyle L. Morris, Jeffrey C. Hoch,
Genji Kurisu, Sameer Velankar, Stephen K. Burley Database (2024) 2024: baae041 https://doi.org/10.1093/database/baae041
The PDB NextGen archive
provides sequence annotation from external resources such as UniProt, SCOP2 and Pfam in addition to the content provided
in the structure model files in the PDB main archive.
The inclusion of UniProtKB numbering facilitates effortless structural comparisons between experimental and
predicted protein models.
These PDBx/mmCIF files are directly compatible with various data visualization tools,
simplifying the display of annotations on 3D structure views.
Structures determined by integrative and hybrid structure determination methods (IHM) are now available alongside
experimental structures in the PDB archive.
These structures are deposited into and processed by the PDB-Dev system.
Each IHM structure is issued a PDB ID, reported in the PDBx/mmCIF file in the _database_2 category, and “integrative” method provenance is captured at _struct.pdbx_structure_determination_methodology.
Users can access and download IHM structures and associated data at files.wwpdb.org/pub/pdb_ihm/.
Currently, holding files in JSON format, validation reports (summary and full reports) in PDF format, and model files in PDBx/mmCIF format are provided.
/pdb_ihm/holdings/ (current holdings, released structures last modified dates, unreleased entries)
/pdb_ihm/data/entries/hash/{PDB_id}/validation_reports/ (includes summary and full validation reports)
/pdb_ihm/data/entries/hash/{PDB_id}/structures/ (latest version of model files)
For example, https://files.wwpdb.org/pub/pdb_ihm/data/entries/zz/8zz1/structures/8zz1.cif.gz
Data may be expanded in the future based on community needs.
In the near future, IHM data will also be available via wwPDB DOI landing pages and wwPDB partner websites.
Congratulations to biocurator Dr. Irina Persikova on processing over 10,000 PDB depositions.
She is the seventh biocurator in the wwPDB reached this milestone.
Irina has received Ph.D. training in Solid State Physics and provided over 20 years of service to the RCSB PDB.
She contributed as an author or co-author to 17 publications and a book chapter.
She has established herself as a highly qualified professional with deep understanding of scientific data
and various experimental techniques and dedication to exceptional quality data curation.
Her profound data curation expertise and commitment to excellence contributed to the high quality data archive
for the benefit of the scientific community.
We congratulate Irina with this exciting accomplishment and look forward to her future success.
As the foundation for depositing, annotating, and archiving structural data across diverse experimental techniques,
the Protein Data Bank Exchange macromolecular Crystallographic Information Framework
(PDBx/mmCIF) stands as the master format of the Protein Data Bank.
Our user-friendly guide offers detailed explanation and examples of essential PDBx/mmCIF records,
aimed to facilitate a smooth transition to this format for depositors and users alike.
The wwPDB anticipates that all four-character PDB IDs will be exhausted by 2028, after which 12-character PDB IDs will be issued.
Entries with extended PDB IDs will not be compatible with the legacy PDB file format and will only be available in PDBx/mmCIF format.
wwPDB encourages users to transition to the PDBx/mmCIF format as soon as possible.
Example PDBx/mmCIF record of a 12-character PDB ID
We invite all users to participate in a brief survey (accessible from the PDBx/mmCIF File Format User Guide) to share feedback on this guide by December 15, 2024. Your feedback will greatly contribute to future developments.
Congratulations to RCSB PDB's Yuhe Liang on processing over 10,000 PDB depositions.
He is the sixth biocurator to reach this milestone in the wwPDB.
Dr. Liang received his PhD in biophysics from Peking University, China with expertise in macromolecular crystallography
and joined the PDB after his postdoctoral training on structural and functional studies of important proteins
related to human health at University of Pittsburgh School of Medicine.
Yuhe Liang
During his 10-year career at RCSB PDB, he has committed his extensive scientific expertise and profound data curation skills
to providing excellent data curation services for the Protein Data Bank.
His dedication and energy has significantly contributed to high quality data archive for the benefit and advancement of the scientific community.
We congratulate Dr. Liang with this exciting accomplishment and look forward to his further career success.
The standardization of protein modification handling ensures that there is a single correct approach to handling
each protein modification that occurs within the PDB archive.
However, there are many existing PDB entries that contain protein modifications which do not follow these handling conventions.
As part of the protein modifications remediation project, all model coordinates files containing protein modifications
are being re-released to add a new protein modification data category.
This new category will list all observed PCMs/PTMs within the entry, as well as their type and category, allowing better findability.
A new category will also be added to the Chemical Component Definition (CCD) files. It will state whether the CCD is a known PCM,
its type and category, as well as on which positions in the amino acid and in the polypeptide it is expected to be observed.
If this PCM is also a known PTM, it will have the Uniprot generic PTM accession ID.
Finally, any protein modifications that are inconsistently handled within a PDB entry will be amended, to ensure that
a given modification is consistently handled in the PDB archive.
The protein chemical modifications (PCMs) and post translational modifications (PTMs) remediation project is
a wwPDB collaborative project carried out principally by PDBe
at EMBL-EBI, and is funded by BBSRC grant number BB/V018779/1.
CASP (Critical Assessment of protein Structure Prediction) is in search for targets.
CASP (Critical Assessment of protein Structure Prediction) experiments are held every two years.
Recent rounds have seen dramatic increases in modeling accuracy, resulting from the introduction of deep learning methods:
In 2018, for the first time, the folds of most proteins were correctly computed [1]; in 2020,
the accuracy of many computed protein structures rivaled that of the corresponding experimental ones [2]; in 2022,
there was an enormous increase in the accuracy of protein complexes [3].
We have seen the beginning of what deep learning methods may achieve in structural biology.
In addition to further increases in the accuracy of protein complexes, methods are being developed for RNA structures,
organic ligand-protein complexes, and for moving beyond single macromolecular structures to compute conformational ensembles.
Accurate computational methods together with experimental data also offer the prospect of probing previously inaccessible biological systems.
CASP has expanded its scope to provide critical assessment in all these areas.
CASP is only possible with the generous participation of the experimental structural biology community in providing suitable targets:
A total of over 1100 targets have been obtained over the previous CASP rounds.
We are now requesting targets for the 2024 CASP16 experiment. We need challenge targets in the following areas:
Single protein structures: The 2020 and 2022 CASPs showed that, so far, Alphafold2 and methods built around it are by far the most accurate [4].
But there are limitations, particularly for some proteins where only a shallow sequence alignment is available and for very large proteins (more than 1000 amino acids).
The best results also require substantial amounts of computing resources, well beyond that of the AlphaFold2 default settings.
Many new methods are continuing to appear and these may remove some of the remaining difficulties.
All types of protein targets are needed, but especially those with shallow sequence alignments, without structural templates, and large proteins.
Protein complexes: In the 2022 CASP15, advanced deep learning methods were applied to protein complexes for the first time [5].
The result was a huge improvement in accuracy compared with classical docking approaches.
But overall, the results are still not at the level achieved for single proteins.
So, in CASP16 we need all sorts of targets in this area so as to determine progress since then.
We particularly need complexes where there is no evolutionary information across the protein-protein interfaces, for example,
antibody-antigen complexes. (This CASP category is conducted in close collaboration with our colleagues at CAPRI
- Critical Assessment of protein interactions [6]).
Nucleic acid structures and complexes: In recognition of the major role nucleic acid structures and complexes play in biology,
CASP now includes this class of target.
A number of papers claiming successful RNA structure computation using deep learning methods have been published,
but those participating in the 2022 CASP RNA category performed less well than classical approaches, and no methods were able to
effectively address the two RNA protein-complexes included [7].
CASP needs a wide variety of RNA, DNA, and complexes as targets to see if this situation has changed.
(This CASP category is conducted in close collaboration with RNApuzzles [8]).
Organic ligand-protein complexes: This area is of major importance for computer-aided drug discovery.
Earlier, there have been community experiments to assess the accuracy of methods, particularly SAMPL, CSAR, D3R, and a new one,
CACHE, has recently started.
These challenges have drawn strong international participation from researchers in both academia and industry.
Here too, a number of promising deep learning papers have appeared, but in the 2022 CASP15 pilot, classical methods were still superior [9].
So, we need appropriate targets to see if progress has been made since.
Ideally, these should be sets of three-dimensional protein-ligand complexes from drug discovery projects,
but single targets would also be appreciated. Additionally, where available, we will assess non-structural quantities such as
affinities or affinity rankings and other properties of pharmaceutical interest when these are available
(small molecule pKs, and DMPK related properties).
Ensembles of macromolecule conformations: It is now widely recognized that proteins and nucleic acids often adopt multiple conformations
that can underpin their functions.
In these cases, considering only a single protein or RNA conformation may be a significant oversimplification.
The 2022 CASP15 included a pilot experiment to assess methods for computing multiple conformations, with encouraging results [10],
but with limitations imposed by the available experimental data.
For 2024, we seek not only cases of multiple experimental three-dimensional structures for the same macromolecule but also
other types of data that might be used for assessment of computed conformation ensembles such as cryoEM, NMR, X-ray crystallography,
SAXS, and/or cross-link data.
Integrative modeling: The more powerful computational methods open up new possibilities for combination with sparse or
low-resolution experimental data to investigate previously inaccessible biological structures and machines.
CASP is interested in exploring these possibilities and so requests experimentally difficult targets where structure has nevertheless been obtained.
In appropriate cases, we expect to be able to collaborate with other experimental groups to provide appropriate data from NMR,
cross-linking or SAXS.
There are three avenues to contribute a target to CASP:
Submit your structure to the PDB (on-hold) and designate it as a CASP target through PDB’s submission interface.
The timeline for the 2024 CASP requires that targets are submitted starting now and until July 1.
We would like to hear from you as soon as possible if you may have something suitable or have suggestions about other target sources.
In order to maintain rigor, the experimental data for a target must not be publicly available until after computed structures have been collected.
For assessment, CASP requires the experimental data by August 15, but the data can remain confidential after that.
Target providers are invited to contribute to papers [11-15] for a special CASP issue of the journal Proteins.
CASP organizers: John Moult, Krzysztof Fidelis, Andriy Kryshtafovych, Torsten Schwede, Maya Topf
Kryshtafovych A, Schwede T, Topf M, Fidelis K, Moult J. Critical assessment of methods of protein structure prediction (CASP)-Round XIII.
Proteins 2019;87(12):1011-1020.
Kryshtafovych A, Schwede T, Topf M, Fidelis K, Moult J. Critical assessment of methods of protein structure prediction (CASP)-Round XIV.
Proteins 2021;89(12):1607-1617.
Kryshtafovych A, Schwede T, Topf M, Fidelis K, Moult J. Critical assessment of methods of protein structure prediction (CASP)-Round XV.
Proteins 2023;91(12):1539-1549.
Ozden B, Kryshtafovych A, Karaca E. The impact of AI-based modeling on the accuracy of protein assembly prediction: Insights from CASP15.
Proteins 2023;91(12):1636-1657.
Lensink MF, Brysbaert G, Raouraoua N, Bates PA, Giulini M, Honorato RV, van Noort C, Teixeira JMC, Bonvin A, Kong R, Shi H, Lu X, Chang S,
Liu J, Guo Z, Chen X, Morehead A, Roy RS, Wu T, Giri N, Quadir F, Chen C, Cheng J, Del Carpio CA, Ichiishi E, Rodriguez-Lumbreras LA,
Fernandez-Recio J, Harmalkar A, Chu LS, Canner S, Smanta R, Gray JJ, Li H, Lin P, He J, Tao H, Huang SY, Roel-Touris J, Jimenez-Garcia B,
Christoffer CW, Jain AJ, Kagaya Y, Kannan H, Nakamura T, Terashi G, Verburgt JC, Zhang Y, Zhang Z, Fujuta H, Sekijima M, Kihara D, Khan O,
Kotelnikov S, Ghani U, Padhorny D, Beglov D, Vajda S, Kozakov D, Negi SS, Ricciardelli T, Barradas-Bautista D, Cao Z, Chawla M, Cavallo L,
Oliva R, Yin R, Cheung M, Guest JD, Lee J, Pierce BG, Shor B, Cohen T, Halfon M, Schneidman-Duhovny D, Zhu S, Yin R, Sun Y, Shen Y,
Maszota-Zieleniak M, Bojarski KK, Lubecka EA, Marcisz M, Danielsson A, Dziadek L, Gaardlos M, Gieldon A, Liwo A, Samsonov SA, Slusarz R,
Zieba K, Sieradzan AK, Czaplewski C, Kobayashi S, Miyakawa Y, Kiyota Y, Takeda-Shitaka M, Olechnovic K, Valancauskas L, Dapkunas J,
Venclovas C, Wallner B, Yang L, Hou C, He X, Guo S, Jiang S, Ma X, Duan R, Qui L, Xu X, Zou X, Velankar S, Wodak SJ.
Impact of AlphaFold on structure prediction of protein complexes: The CASP15-CAPRI experiment.
Proteins 2023;91(12):1658-1683.
Das R, Kretsch RC, Simpkin AJ, Mulvaney T, Pham P, Rangan R, Bu F, Keegan RM, Topf M, Rigden DJ, Miao Z, Westhof E.
Assessment of three-dimensional RNA structure prediction in CASP15.
Proteins 2023;91(12):1747-1770.
Magnus M, Antczak M, Zok T, Wiedemann J, Lukasiak P, Cao Y, Bujnicki JM, Westhof E, Szachniuk M, Miao Z. RNA-Puzzles toolkit:
a computational resource of RNA 3D structure benchmark datasets, structure manipulation, and evaluation tools.
Nucleic Acids Res 2020;48(2):576-588.
Robin X, Studer G, Durairaj J, Eberhardt J, Schwede T, Walters WP. Assessment of protein-ligand complexes in CASP15.
Proteins 2023;91(12):1811-1821.
Kryshtafovych A, Montelione GT, Rigden DJ, Mesdaghi S, Karaca E, Moult J. Breaking the conformational ensemble barrier:
Ensemble structure modeling challenges in CASP15.
Proteins 2023;91(12):1903-1911.
Kretsch RC, Andersen ES, Bujnicki JM, Chiu W, Das R, Luo B, Masquida B, McRae EKS, Schroeder GM, Su Z, Wedekind JE, Xu L,
Zhang K, Zheludev IN, Moult J, Kryshtafovych A. RNA target highlights in CASP15: Evaluation of predicted models by structure providers.
Proteins 2023;91(12):1600-1615.
Alexander LT, Durairaj J, Kryshtafovych A, Abriata LA, Bayo Y, Bhabha G, Breyton C, Caulton SG, Chen J, Degroux S, Ekiert DC, Erlandsen BS,
Freddolino PL, Gilzer D, Greening C, Grimes JM, Grinter R, Gurusaran M, Hartmann MD, Hitchman CJ, Keown JR, Kropp A, Kursula P, Lovering AL,
Lemaitre B, Lia A, Liu S, Logotheti M, Lu S, Markusson S, Miller MD, Minasov G, Niemann HH, Opazo F, Phillips GN, Jr., Davies OR,
Rommelaere S, Rosas-Lemus M, Roversi P, Satchell K, Smith N, Wilson MA, Wu KL, Xia X, Xiao H, Zhang W, Zhou ZH, Fidelis K, Topf M, Moult J,
Schwede T. Protein target highlights in CASP15: Analysis of models by structure providers.
Proteins 2023;91(12):1571-1599.
Alexander LT, Lepore R, Kryshtafovych A, Adamopoulos A, Alahuhta M, Arvin AM, Bomble YJ, Bottcher B, Breyton C, Chiarini V, Chinnam NB,
Chiu W, Fidelis K, Grinter R, Gupta GD, Hartmann MD, Hayes CS, Heidebrecht T, Ilari A, Joachimiak A, Kim Y, Linares R, Lovering AL,
Lunin VV, Lupas AN, Makbul C, Michalska K, Moult J, Mukherjee PK, Nutt WS, Oliver SL, Perrakis A, Stols L, Tainer JA, Topf M, Tsutakawa SE,
Valdivia-Delgado M, Schwede T. Target highlights in CASP14: Analysis of models by structure providers.
Proteins 2021;89(12):1647-1672.
Lepore R, Kryshtafovych A, Alahuhta M, Veraszto HA, Bomble YJ, Bufton JC, Bullock AN, Caba C, Cao H, Davies OR, Desfosses A, Dunne M,
Fidelis K, Goulding CW, Gurusaran M, Gutsche I, Harding CJ, Hartmann MD, Hayes CS, Joachimiak A, Leiman PG, Loppnau P, Lovering AL,
Lunin VV, Michalska K, Mir-Sanchis I, Mitra AK, Moult J, Phillips GN, Jr., Pinkas DM, Rice PA, Tong Y, Topf M, Walton JD, Schwede T.
Target highlights in CASP13: Experimental target structures through the eyes of their authors.
Proteins 2019;87(12):1037-1057.
Kryshtafovych A, Albrecht R, Basle A, Bule P, Caputo AT, Carvalho AL, Chao KL, Diskin R, Fidelis K, Fontes C, Fredslund F, Gilbert HJ,
Goulding CW, Hartmann MD, Hayes CS, Herzberg O, Hill JC, Joachimiak A, Kohring GW, Koning RI, Lo Leggio L, Mangiagalli M, Michalska K,
Moult J, Najmudin S, Nardini M, Nardone V, Ndeh D, Nguyen TH, Pintacuda G, Postel S, van Raaij MJ, Roversi P, Shimon A, Singh AK,
Sundberg EJ, Tars K, Zitzmann N, Schwede T. Target highlights from the first post-PSI CASP experiment (CASP12, May-August 2016).
Proteins 2018;86 Suppl 1(Suppl 1):27-50.
Mechanisms of Light Signalling and Allosteric Regulation in Dual Sensor Photoreceptor PPHK
Irin Pottanani Tom (1), Heewhan Shin (1), Chang Liu (2), Indika Kumaeapperuma (1), Zhong Ren (1), Minglei Zhao (1),
Xiaojing Yang (1) 1) University of Illinois at Chicago, 2) University of Chicago
Many thanks to The Biophysical Society organizers and poster prize judges for making this award possible.
The wwPDB Foundation was established in 2010 to raise funds
in support of the outreach activities of the wwPDB.
The Foundation raised funds to help support PDB50 events, workshops, and educational publications.
The Foundation is chartered as a 501(c)(3) entity exclusively for scientific, literary, charitable, and educational purposes.
Congratulations to biocurator Minyu Chen on processing over 10,000 PDB depositions.
She is the second biocurator to reach this milestone in the PDBj and the fifth in the wwPDB.
Yumiko Kengaku reached this milestone in April 2021.
Minyu received her PhD in Environmental Engineering from Osaka University and joined PDB
after working at the National Cerebral and Cardiovascular Center, Osaka.
She has joined PDB in 2007 and is now working at the branch office of PDBj in the Protein Research Foundation, Osaka.
She has established herself as a highly qualified professional with deep understanding of scientific data
and various experimental techniques and dedication to exceptional quality data curation.
Her profound data curation expertise and commitment to excellence contributed to the high quality data archive
for the benefit of the scientific community.
We congratulate Minyu with this exciting accomplishment and look forward to her future success.
Chairman of the Protein Research Foundation, Prof. Toshiharu Hase, and Dr. Minyu Chen.
[wwPDB] Preprint Published on NMR Restraint Validation
Graphical Abstract
This manuscript addresses this challenge of validation of experimental biomolecular NMR structures against restraint data.
The NMR exchange (NEF) and NMR-STAR formats provide a standardized approach for representing commonly used NMR restraints.
Using these restraint formats, a standardized validation system for assessing structural models of biopolymers
against restraints has been developed and implemented in the wwPDB OneDep data harvesting system.
The resulting wwPDB Restraint Violation Report provides a model vs data assessment of biomolecule structures
determined using distance and dihedral restraints, with extensions to other restraint types currently
being implemented. These tools are useful for assessing NMR models,
as well as for assessing biomolecular structure predictions based on distance restraints.
We presented the rationale for model-vs-data restraint validation by the wwPDB,
together with summary of validation tools and reports for NMR distance and dihedral restraints
that have been developed, as implemented in the wwPDB validation pipeline and recommended
by the wwPDB NMR-VTF committee.
Restraint Validation of Biomolecular Structures Determined by NMR in the Protein Data Bank
Kumaran Baskaran, Eliza Ploskon, Roberto Tejero, Masashi Yokochi, Deborah Harrus, Yuhe Liang, Ezra Peisach,
Irina Persikova, Theresa A Ramelot, Monica Sekharan, James Tolchard, John D Westbrook, Benjamin Bardiaux,
Charles Schwieters, Ardan Patwardhan, Sameer Velankar, Stephen K Burley, Genji Kurisu,
Jeffrey C Hoch, Gaetano T Montelione, Geerten W Vuister, Jasmine Y Young
(2024) bioRxiv 2024.01.15.575520;
doi: 10.1101/2024.01.15.575520
wwPDB plans to further enhance validation report by providing model-vs-data quality assessment
for other kinds of restraints based on community recommendation and improve data representation
on structures with multiple conformation states.
[wwPDB] Preprint Published on CryoEM Archiving and Validation Recommendations
The number of released EMDB entries per year in a number of resolution bins, from 2010 until December 2023
A workshop was held at EMBL-EBI (Hinxton, UK) in January 2020 to discuss data requirements for deposition
and validation of cryoEM structures, with a focus on single-particle analysis and set community recommendations.
Community recommendations on cryoEM data archiving and validation
Gerard J. Kleywegt, Paul D. Adams, Sarah J. Butcher, Cathy Lawson, Alexis Rohou, Peter B. Rosenthal,
Sriram Subramaniam, Maya Topf, Sanja Abbott, Philip R. Baldwin, John M. Berrisford, Gérard Bricogne,
Preeti Choudhary, Tristan I. Croll, Radostin Danev, Sai J. Ganesan, Timothy Grant, Aleksandras Gutmanas,
Richard Henderson, J. Bernard Heymann, Juha T. Huiskonen, Andrei Istrate, Takayuki Kato, Gabriel C. Lander,
Shee-Mei Lok, Steven J. Ludtke, Garib N. Murshudov, Ryan Pye, Grigore D. Pintilie, Jane S. Richardson,
Carsten Sachse, Osman Salih, Sjors H.W. Scheres, Gunnar F. Schroeder, Carlos Oscar S. Sorzano,
Scott M. Stagg, Zhe Wang, Rangana Warshamanage, John D. Westbrook, Martyn D. Winn, Jasmine Y. Young,
Stephen K. Burley, Jeffrey C. Hoch, Genji Kurisu, Kyle Morris, Ardan Patwardhan, Sameer Velankar
(2023) arXiv doi: 10.48550/arXiv.2311.17640
Several community recommendations from this workshop have been incorporated into wwPDB validation reports
including map analysis, FSC validation, and map-model fitness using Q-score.
wwPDB plans to provide overall quality percentile on map-model fitness compared to other PDB entries
in the wwPDB validation report as the next step.
A new paper describes how the
recently-announced NextGen Archive
provides centralized access to integrated annotations and enriched structural information for PDB data:
NextGen Archive: Centralising Access to Integrated Annotations and Enriched Structural Information
by the Worldwide Protein Data Bank
Preeti Choudhary, Zukang Feng, John Berrisford, Henry Chao, Yasuyo Ikegawa, Ezra Peisach, Dennis W. Piehl,
James Smith, Ahsan Tanweer, Mihaly Varadi, John D. Westbrook, Jasmine Y. Young, Ardan Patwardhan,
Kyle L. Morris, Jeffrey C. Hoch, Genji Kurisu, Sameer Velankar, Stephen K. Burley
(2023) bioRxiv doi: 10.1101/2023.10.24.563739
The PDB NextGen archive
provides sequence annotation from external resources such as UniProt, SCOP2 and Pfam
in addition to the content provided in the structure model files in the PDB main archive.
The inclusion of UniProtKB numbering facilitates effortless structural comparisons
between experimental and predicted protein models.
These PDBx/mmCIF files are directly compatible with various data visualization tools,
simplifying the display of annotations on 3D structure views.
Thermodynamic analysis of Fv-supercharged antibody–antigen interactions and control of interaction parameters
Keisuke Kasahara (1), Daisuke Kuroda (2), Jose Caaveiro (3), Satoru Nagatoishi (4), Kouhei Tsumoto (1,4)
1) Dept. Bioeng., Grad. Sch. Eng., Univ. Tokyo; 2) Res. Ctr. Drug Vaccine Dev., NIID;
3) Grad. Sch. Pharm. Sci., Kyusyu Univ., 4) Med. Dev. Dev. Reg. Res. Ctr., Grad. Sch. Eng., Univ. Tokyo
Kyle Ian Peter Le Huray
Harnessing the power of machine learning and high-throughput molecular dynamics simulations to predict protein-lipid interactions Kyle Ian Peter Le Huray (1,2), Frank Sobott (1), He Wang (3), Antreas Kalli (2) 1) School of Molecular and Cellular Biology, Faculty of Biological Sciences, University of Leeds, Leeds, UK; 2) Leeds Institute of Cardiovascular and Metabolic Medicine, School of Medicine, University of Leeds, Leeds, UK; 3) School of Computing, University of Leeds, Leeds, UK
Katsuhiko Minami
Replication-dependent histone (Repli-Histo) labeling revealed that chromatin motion can determine DNA replication timing
Katsuhiko Minami (1,2), Satoru Ide (1,2), Sachiko Tamura (1), Masato T. Kanemaki (1,2), Kazuhiro Maeshima (1,2)
1) National Institute of Genetics; 2) Graduate Institute for Advanced Studies, SOKENDAI
Many thanks to the meeting organizers and prize judges for making these awards possible.
The wwPDB Foundation was established in 2010
to raise funds in support of the outreach activities of the wwPDB.
The Foundation raised funds to help support PDB50 events, workshops, and educational publications.
The Foundation is chartered as a 501(c)(3) entity exclusively for scientific, literary, charitable, and educational purposes.
Consider supporting the next 50 years of PDB's spirit of openness, cooperation, and education with a
donation to the wwPDB Foundation.
wwPDB anticipates that all the four character PDB accession codes (PDB ID) will be consumed by 2029.
With the continuous growth of PDB archive, wwPDB has revised the PDB accession code format
by extending its length and prepending “PDB” (e.g., "1abc" will become "pdb_00001abc").
This process will enable text mining detection of PDB entries in the published literature
and allow for more informative and transparent delivery of revised data files.
Entries with extended PDB IDs (12 characters) will not be compatible with the legacy PDB file format
once four-character PDB IDs are consumed.
wwPDB encourages scientific journals, PDB community and users to transition to using the PDBx/mmCIF format
and the extended PDB ID format as soon as possible.
Resources are available to help PDB users with this transition through the
wwPDB resource portal page (Extended PDB ID With 12 Characters).
This page links to useful resources for handling this change, including an
FAQ on PDB ID extension, materials to learn more about
PDBx/mmCIF format, and links to other PDBx/mmCIF resources and software tools.
As the transition phase progresses, more training resources will be added to this page.
Additionally, a PDB “beta” archive will be provided during the transition phase in 2026.
The directory structure of this “beta” archive will mirror the data organization of the
PDB Versioned Archive in the form of
https://files-beta.org/pub/pdb/data/entries/
two-letter-hash/pdb_accession_code/entry_data_File_names.
The two-letter hash will be based on the n-2 and n-3 characters.
For example, PDB entry PDB_12345678 will be under /67/.
This will maintain consistency with the current PDB archive, where e.g. PDB entry 1abc is under /ab.
Once all the four character PDB accession codes are consumed, this PDB “beta” archive
will become the PDB main archive and the current PDB archive will be removed.
Download example files containing extended PDB IDs for software adoption
from GitHub.
The directory 20240101 includes the 214,121 experimentally-determined structure and experimental data available at that time.
Atomic coordinate and related metadata are available in PDBx/mmCIF, PDB, and XML file formats.
The date and time stamp of each file indicates the last time the file was modified.
The snapshot of PDB Core Archive is 1,242 GB.

")

 Integrative structures are available at wwPDB.org and the PDB archive
Integrative structures are available at wwPDB.org and the PDB archive


 Irina Persikova
Irina Persikova






 Chairman of the Protein Research Foundation, Prof. Toshiharu Hase, and Dr. Minyu Chen.
Chairman of the Protein Research Foundation, Prof. Toshiharu Hase, and Dr. Minyu Chen.
 Milestone tumbler.
Milestone tumbler.
 Graphical Abstract
Graphical Abstract



 Katsuhiko Minami
Katsuhiko Minami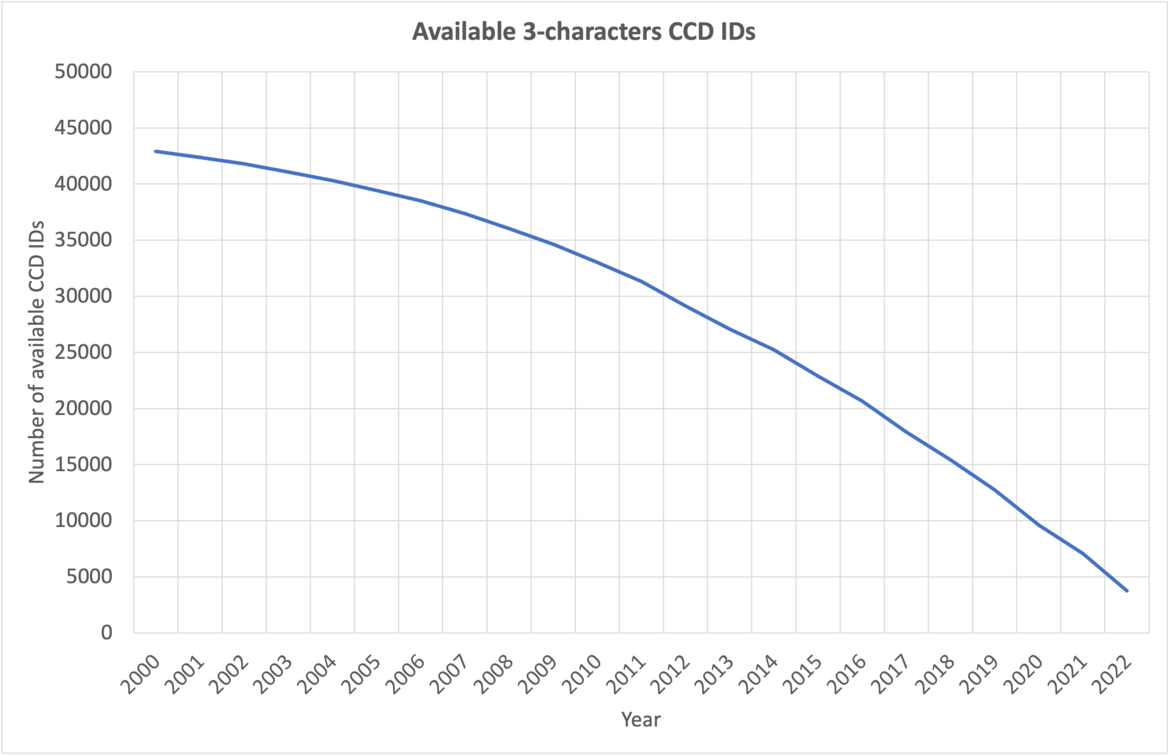 The number of available 3-character CCD IDs annually.
The number of available 3-character CCD IDs annually.




