Journal: Nat Commun / Year: 2019 Title: Single particle cryo-EM reconstruction of 52 kDa streptavidin at 3.2 Angstrom resolution. Authors: Xiao Fan / Jia Wang / Xing Zhang / Zi Yang / Jin-Can Zhang / Lingyun Zhao / Hai-Lin Peng / Jianlin Lei / Hong-Wei Wang / Abstract: The fast development of single-particle cryogenic electron microscopy (cryo-EM) has made it more feasible to obtain the 3D structure of well-behaved macromolecules with a molecular weight higher than ...The fast development of single-particle cryogenic electron microscopy (cryo-EM) has made it more feasible to obtain the 3D structure of well-behaved macromolecules with a molecular weight higher than 300 kDa at ~3 Å resolution. However, it remains a challenge to obtain the high-resolution structures of molecules smaller than 200 kDa using single-particle cryo-EM. In this work, we apply the Cs-corrector-VPP-coupled cryo-EM to study the 52 kDa streptavidin (SA) protein supported on a thin layer of graphene and embedded in vitreous ice. We are able to solve both the apo-SA and biotin-bound SA structures at near-atomic resolution using single-particle cryo-EM. We demonstrate that the method has the potential to determine the structures of molecules as small as 39 kDa.
History
Deposition
Jan 15, 2019
Deposition site: PDBJ / Processing site: PDBJ
Revision 1.0
May 29, 2019
Provider: repository / Type: Initial release
Revision 1.0
May 29, 2019
Data content type: EM metadata / Data content type: EM metadata / Provider: repository / Type: Initial release
Revision 1.0
May 29, 2019
Data content type: FSC / Data content type: FSC / Provider: repository / Type: Initial release
Revision 1.0
May 29, 2019
Data content type: Image / Data content type: Image / Provider: repository / Type: Initial release
Revision 1.0
May 29, 2019
Data content type: Primary map / Data content type: Primary map / Provider: repository / Type: Initial release
Revision 1.0
May 29, 2019
Data content type: FSC / Data content type: FSC / Provider: repository / Type: Initial release
Revision 1.0
May 29, 2019
Data content type: Image / Data content type: Image / Provider: repository / Type: Initial release
Revision 1.0
May 29, 2019
Data content type: Primary map / Data content type: Primary map / Provider: repository / Type: Initial release
Revision 1.0
May 29, 2019
Data content type: FSC / Data content type: FSC / Provider: repository / Type: Initial release
Revision 1.0
May 29, 2019
Data content type: Image / Data content type: Image / Provider: repository / Type: Initial release
Revision 1.0
May 29, 2019
Data content type: Primary map / Data content type: Primary map / Provider: repository / Type: Initial release
Revision 1.0
May 29, 2019
Data content type: FSC / Data content type: FSC / Provider: repository / Type: Initial release
Revision 1.0
May 29, 2019
Data content type: Image / Data content type: Image / Provider: repository / Type: Initial release
Revision 1.0
May 29, 2019
Data content type: Primary map / Data content type: Primary map / Provider: repository / Type: Initial release
Revision 1.0
May 29, 2019
Data content type: FSC / Data content type: FSC / Provider: repository / Type: Initial release
Revision 1.0
May 29, 2019
Data content type: Image / Data content type: Image / Provider: repository / Type: Initial release
Revision 1.0
May 29, 2019
Data content type: Primary map / Data content type: Primary map / Provider: repository / Type: Initial release
Data content type: EM metadata / Data content type: EM metadata / EM metadata / Group: Data processing / Experimental summary / Data content type: EM metadata / EM metadata / Category: em_admin / em_software / Data content type: EM metadata / EM metadata / Item: _em_admin.last_update / _em_software.name
EMPIAR-10270 (Title: Single particle reconstruction of 52 kDa biotin-bound state streptavidin at 3.2 Angstrom resolution Data size: 5.5 TB Data #1: Uncorrected biotinbound state streptavidin movie stacks, binning 2 from super-resolution stacks. [micrographs - multiframe])
Average exposure time: 2.56 sec. / Electron dose: 50 e/Å2 / Detector mode: SUPER-RESOLUTION / Film or detector model: GATAN K2 SUMMIT (4k x 4k) / Num. of grids imaged: 1 / Num. of real images: 1450
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information Streptomyces avidinii (bacteria)
Streptomyces avidinii (bacteria) Authors
Authors China, 1items
China, 1items  Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads












 PDBj
PDBj Assembly
Assembly
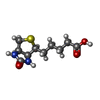
 Mass: 244.311 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C10H16N2O3S
Mass: 244.311 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C10H16N2O3S Mass: 18.015 Da / Num. of mol.: 12 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 12 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation Electron microscopy imaging
Electron microscopy imaging
 FIELD EMISSION GUN / Accelerating voltage: 300 kV / Illumination mode: FLOOD BEAM
FIELD EMISSION GUN / Accelerating voltage: 300 kV / Illumination mode: FLOOD BEAM Processing
Processing