 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-6ibl: ACTIVATED TURKEY BETA1 ADRENOCEPTOR WITH BOUND AGONIST FORMOTEROL... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 6ibl | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | ACTIVATED TURKEY BETA1 ADRENOCEPTOR WITH BOUND AGONIST FORMOTEROL AND NANOBODY Nb80 | |||||||||
 Components Components |
| |||||||||
 Keywords Keywords | IMMUNE SYSTEM / Beta1 Adrenoceptor / Activated / Agonist / Nanobody | |||||||||
| Function / homology |  Function and homology information Function and homology informationbeta1-adrenergic receptor activity / positive regulation of heart contraction / regulation of circadian sleep/wake cycle, sleep / norepinephrine-epinephrine-mediated vasodilation involved in regulation of systemic arterial blood pressure / DNA polymerase processivity factor activity / protein-disulfide reductase activity / adenylate cyclase-activating adrenergic receptor signaling pathway / cell redox homeostasis / early endosome / positive regulation of MAPK cascade ...beta1-adrenergic receptor activity / positive regulation of heart contraction / regulation of circadian sleep/wake cycle, sleep / norepinephrine-epinephrine-mediated vasodilation involved in regulation of systemic arterial blood pressure / DNA polymerase processivity factor activity / protein-disulfide reductase activity / adenylate cyclase-activating adrenergic receptor signaling pathway / cell redox homeostasis / early endosome / positive regulation of MAPK cascade / identical protein binding / membrane / plasma membrane / cytosol / cytoplasm Similarity search - Function | |||||||||
| Biological species |    | |||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.7 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.7 Å | |||||||||
 Authors Authors | Warne, T. / Edwards, P.C. / Dore, A.S. / Leslie, A.G.W. / Tate, C.G. | |||||||||
 Citation Citation | Journal: Nature / Year: 2020 Title: Molecular basis of β-arrestin coupling to formoterol-bound β-adrenoceptor. Authors: Yang Lee / Tony Warne / Rony Nehmé / Shubhi Pandey / Hemlata Dwivedi-Agnihotri / Madhu Chaturvedi / Patricia C Edwards / Javier García-Nafría / Andrew G W Leslie / Arun K Shukla / Christopher G Tate /     Abstract: The β-adrenoceptor (βAR) is a G-protein-coupled receptor (GPCR) that couples to the heterotrimeric G protein G. G-protein-mediated signalling is terminated by phosphorylation of the C terminus of ...The β-adrenoceptor (βAR) is a G-protein-coupled receptor (GPCR) that couples to the heterotrimeric G protein G. G-protein-mediated signalling is terminated by phosphorylation of the C terminus of the receptor by GPCR kinases (GRKs) and by coupling of β-arrestin 1 (βarr1, also known as arrestin 2), which displaces G and induces signalling through the MAP kinase pathway. The ability of synthetic agonists to induce signalling preferentially through either G proteins or arrestins-known as biased agonism-is important in drug development, because the therapeutic effect may arise from only one signalling cascade, whereas the other pathway may mediate undesirable side effects. To understand the molecular basis for arrestin coupling, here we determined the cryo-electron microscopy structure of the βAR-βarr1 complex in lipid nanodiscs bound to the biased agonist formoterol, and the crystal structure of formoterol-bound βAR coupled to the G-protein-mimetic nanobody Nb80. βarr1 couples to βAR in a manner distinct to that of G coupling to βAR-the finger loop of βarr1 occupies a narrower cleft on the intracellular surface, and is closer to transmembrane helix H7 of the receptor when compared with the C-terminal α5 helix of G. The conformation of the finger loop in βarr1 is different from that adopted by the finger loop of visual arrestin when it couples to rhodopsin. βAR coupled to βarr1 shows considerable differences in structure compared with βAR coupled to Nb80, including an inward movement of extracellular loop 3 and the cytoplasmic ends of H5 and H6. We observe weakened interactions between formoterol and two serine residues in H5 at the orthosteric binding site of βAR, and find that formoterol has a lower affinity for the βAR-βarr1 complex than for the βAR-G complex. The structural differences between these complexes of βAR provide a foundation for the design of small molecules that could bias signalling in the β-adrenoceptors. #1: Journal: Biorxiv / Year: 2018Title: Molecular basis for high affinity agonist binding in GPCRs Authors: Warne, T. / Edwards, P.C. / Dore, A.S. / Leslie, A.G.W. / Tate, C.G. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 6ibl.cif.gz | 220.9 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb6ibl.ent.gz | 175.2 KB | Display | PDB format |
| PDBx/mmJSON format | 6ibl.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ib/6iblftp://data.pdbj.org/pub/pdb/validation_reports/ib/6ibl | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  6tkoC  2h6xS  3p0gS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |











-Links
 PDBj
PDBj











- Assembly
Assembly
| Deposited unit | 
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | 
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Unit cell |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Noncrystallographic symmetry (NCS) | NCS domain:
NCS domain segments:
NCS ensembles :
|
-Components
-Protein / Antibody , 2 types, 4 molecules ABCD
| #1: Protein | Mass: 46769.094 Da / Num. of mol.: 2 / Mutation: C32S,C35S,C32S,C35S Source method: isolated from a genetically manipulated source Source: (gene. exp.) Strain: K12 / Gene: trxA, fipA, tsnC, b3781, JW5856, ADRB1 / Production host:  Trichoplusia ni (cabbage looper) / References: UniProt: P0AA25, UniProt: P07700 Trichoplusia ni (cabbage looper) / References: UniProt: P0AA25, UniProt: P07700#2: Antibody | Mass: 13042.373 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) |
|---|
-Non-polymers , 4 types, 41 molecules 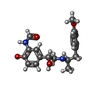

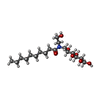

| #3: Chemical |  Mass: 344.405 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C19H24N2O4 / Comment: agonist*YM Mass: 344.405 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C19H24N2O4 / Comment: agonist*YM#4: Chemical |  Mass: 22.990 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Na Mass: 22.990 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Na#5: Chemical | ChemComp-2CV /  Mass: 379.489 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: C18H37NO7 / Comment: detergent*YM Mass: 379.489 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: C18H37NO7 / Comment: detergent*YM#6: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 29 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Details
| Has protein modification | Y |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 4.02 Å3/Da / Density % sol: 69.41 % |
|---|---|
| Crystal grow | Temperature: 291 K / Method: vapor diffusion, sitting drop / pH: 7.5 / Details: 0.1 M Hepes-NaOH pH7.5 and 21-24% PEG1500 |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: MASSIF-1 / Wavelength: 0.966 Å / Beamline: MASSIF-1 / Wavelength: 0.966 Å |
| Detector | Type: DECTRIS PILATUS3 X 2M / Detector: PIXEL / Date: Dec 15, 2015 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.966 Å / Relative weight: 1 |
| Reflection | Resolution: 2.7→88.57 Å / Num. obs: 50611 / % possible obs: 99 % / Redundancy: 4.6 % / Net I/σ(I): 6.6 |
| Reflection shell | Resolution: 2.7→2.79 Å |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 2H6X, 3P0G Resolution: 2.7→44.32 Å / Cor.coef. Fo:Fc: 0.856 / Cor.coef. Fo:Fc free: 0.786 / SU B: 16.666 / SU ML: 0.326 / Cross valid method: THROUGHOUT / ESU R Free: 0.464 / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 66.667 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: 1 / Resolution: 2.7→44.32 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|