National Institutes of Health/National Institute of General Medical Sciences (NIH/NIGMS)
R01GM065546
United States
Citation
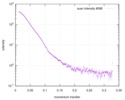
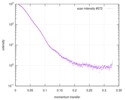
Journal: FEBS J / Year: 2017 Title: Biophysical investigation of type A PutAs reveals a conserved core oligomeric structure. Authors: David A Korasick / Harkewal Singh / Travis A Pemberton / Min Luo / Richa Dhatwalia / John J Tanner / Abstract: Many enzymes form homooligomers, yet the functional significance of self-association is seldom obvious. Herein, we examine the connection between oligomerization and catalytic function for proline ...Many enzymes form homooligomers, yet the functional significance of self-association is seldom obvious. Herein, we examine the connection between oligomerization and catalytic function for proline utilization A (PutA) enzymes. PutAs are bifunctional enzymes that catalyze both reactions of proline catabolism. Type A PutAs are the smallest members of the family, possessing a minimal domain architecture consisting of N-terminal proline dehydrogenase and C-terminal l-glutamate-γ-semialdehyde dehydrogenase modules. Type A PutAs form domain-swapped dimers, and in one case (Bradyrhizobium japonicum PutA), two of the dimers assemble into a ring-shaped tetramer. Whereas the dimer has a clear role in substrate channeling, the functional significance of the tetramer is unknown. To address this question, we performed structural studies of four-type A PutAs from two clades of the PutA tree. The crystal structure of Bdellovibrio bacteriovorus PutA covalently inactivated by N-propargylglycine revealed a fold and substrate-channeling tunnel similar to other PutAs. Small-angle X-ray scattering (SAXS) and analytical ultracentrifugation indicated that Bdellovibrio PutA is dimeric in solution, in contrast to the prediction from crystal packing of a stable tetrameric assembly. SAXS studies of two other type A PutAs from separate clades also suggested that the dimer predominates in solution. To assess whether the tetramer of B. japonicum PutA is necessary for catalytic function, a hot spot disruption mutant that cleanly produces dimeric protein was generated. The dimeric variant exhibited kinetic parameters similar to the wild-type enzyme. These results implicate the domain-swapped dimer as the core structural and functional unit of type A PutAs. ENZYMES: Proline dehydrogenase (EC 1.5.5.2); l-glutamate-γ-semialdehyde dehydrogenase (EC 1.2.1.88). DATABASES: The atomic coordinates and structure factor amplitudes have been deposited in the Protein Data Bank under accession number 5UR2. The SAXS data have been deposited in the SASBDB under the ...DATABASES: The atomic coordinates and structure factor amplitudes have been deposited in the Protein Data Bank under accession number 5UR2. The SAXS data have been deposited in the SASBDB under the following accession codes: SASDCP3 (BbPutA), SASDCQ3 (DvPutA 1.5 mg·mL ), SASDCX3 (DvPutA 3.0 mg·mL ), SASDCY3 (DvPutA 4.5 mg·mL ), SASDCR3 (LpPutA 3.0 mg·mL ), SASDCV3 (LpPutA 5.0 mg·mL ), SASDCW3 (LpPutA 8.0 mg·mL ), SASDCS3 (BjPutA 2.3 mg·mL ), SASDCT3 (BjPutA 4.7 mg·mL ), SASDCU3 (BjPutA 7.0 mg·mL ), SASDCZ3 (R51E 2.3 mg·mL ), SASDC24 (R51E 4.7 mg·mL ), SASDC34 (R51E 7.0 mg·mL ).
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information Bdellovibrio bacteriovorus (bacteria)
Bdellovibrio bacteriovorus (bacteria) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors United States, 1items
United States, 1items  Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads



















 PDBj
PDBj

 Assembly
Assembly




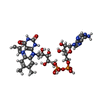
 Mass: 898.664 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C32H40N10O17P2
Mass: 898.664 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C32H40N10O17P2 Mass: 18.015 Da / Num. of mol.: 1048 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 1048 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation Processing
Processing