 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 5d9z | ||||||
|---|---|---|---|---|---|---|---|
| Title | Structure of Colocasia Esculenta Agglutinin with mannose bound | ||||||
 Components Components | (Tuber agglutinin) x 2 | ||||||
 Keywords Keywords | SUGAR BINDING PROTEIN / LECTIN / PROTEIN-CARBOHYDRATE INTERACTIONS / DIETARY PROTEIN / BETA PRISM II FOLD / 18-AUG | ||||||
| Function / homology |  Function and homology information Function and homology informationresponse to other organism / D-mannose binding / extracellular region / metal ion binding Similarity search - Function | ||||||
| Biological species |  Colocasia esculenta (taro) Colocasia esculenta (taro) | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 1.85 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 1.85 Å | ||||||
 Authors Authors | Chattopadhyaya, R. | ||||||
| Funding support |  India, 1items India, 1items
| ||||||
 Citation Citation | Journal: to be published Title: High resolution crystal structures of Colocasia esculenta agglutinin with and without mannose Authors: Chattopadhyaya, R. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 5d9z.cif.gz | 60 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb5d9z.ent.gz | 42.5 KB | Display | PDB format |
| PDBx/mmJSON format | 5d9z.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/d9/5d9zftp://data.pdbj.org/pub/pdb/validation_reports/d9/5d9z | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  3r0eS S: Starting model for refinement |
|---|---|
| Similar structure data |











-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Components on special symmetry positions |
|
-Components
| #1: Protein | Mass: 12012.415 Da / Num. of mol.: 1 / Fragment: UNP residues 24-132 / Source method: isolated from a natural source / Source: (natural) Colocasia esculenta (taro) / Plasmid details: local market, food item / Tissue: tuber / References: UniProt: R9RL27 | ||||||
|---|---|---|---|---|---|---|---|
| #2: Protein | Mass: 12489.940 Da / Num. of mol.: 1 / Fragment: UNP residues 140-251 / Source method: isolated from a natural source / Source: (natural) Colocasia esculenta (taro) / Plasmid details: local market, food item / Tissue: tuber / References: UniProt: R9RL27 | ||||||
| #3: Sugar | ChemComp-BMA / 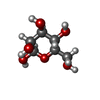  Type: D-saccharide, beta linking / Mass: 180.156 Da / Num. of mol.: 5 Type: D-saccharide, beta linking / Mass: 180.156 Da / Num. of mol.: 5Source method: isolated from a genetically manipulated source Formula: C6H12O6 #4: Chemical | ChemComp-PO4 / |   Mass: 94.971 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: PO4 Mass: 94.971 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: PO4#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 33 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 33 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 4.23 Å3/Da / Density % sol: 70.89 % / Description: resembles a piece of cut diamond |
|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion, hanging drop / pH: 5.6 Details: 50 molar excess of D-Mannose was added to the purified 8.10 mg/ml lectin stock solution; 2 vols of this was mixed with 1 vol of crystallizing agent containing 0.1 M sodium citrate tribasic ...Details: 50 molar excess of D-Mannose was added to the purified 8.10 mg/ml lectin stock solution; 2 vols of this was mixed with 1 vol of crystallizing agent containing 0.1 M sodium citrate tribasic pH 5.6 and 1.0 M ammonium phosphate monobasic; equilibrated with above crystallizing agent in reservoir; large crystals in 3 weeks Temp details: fluctuated within 5K |
-Data collection
| Diffraction | Mean temperature: 111 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RUH3R / Wavelength: 1.5419 Å |
| Detector | Type: RIGAKU RAXIS IV++ / Detector: IMAGE PLATE / Date: Dec 7, 2014 |
| Radiation | Monochromator: Ni filter / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5419 Å / Relative weight: 1 |
| Reflection | Resolution: 1.7→29.04 Å / Num. obs: 39501 / % possible obs: 85.6 % / Redundancy: 9.13 % / Rmerge(I) obs: 0.313 / Net I/σ(I): 4 |
| Reflection shell | Resolution: 1.7→1.76 Å / Redundancy: 3.62 % / Rmerge(I) obs: 0.664 / Mean I/σ(I) obs: 1.3 / % possible all: 24.7 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 3R0E Resolution: 1.85→19.803 Å / SU ML: 0.7 / Cross valid method: FREE R-VALUE / σ(F): 1.37 / Phase error: 60.47 / Stereochemistry target values: ML
| ||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL | ||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.85→19.803 Å
| ||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION
|
