 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 5t1x | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Title | Crystal Structure of Native Tarin Lectin | ||||||||||||
 Components Components | (Lectin) x 2 | ||||||||||||
 Keywords Keywords | SUGAR BINDING PROTEIN / Tarin / GNA-related Lectin | ||||||||||||
| Function / homology |  Function and homology information Function and homology information | ||||||||||||
| Biological species |  Colocasia esculenta (taro) Colocasia esculenta (taro) | ||||||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.7 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.7 Å | ||||||||||||
 Authors Authors | Pereira, P.R. / Meagher, J.L. / Stuckey, J.A. | ||||||||||||
| Funding support |  Brazil, Brazil,  United States, 3items United States, 3items
| ||||||||||||
 Citation Citation | Journal: Glycobiology / Year: 2017 Title: High-resolution crystal structures of Colocasia esculenta tarin lectin. Authors: Pereira, P.R. / Meagher, J.L. / Winter, H.C. / Goldstein, I.J. / Paschoalin, V.M. / Silva, J.T. / Stuckey, J.A. | ||||||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 5t1x.cif.gz | 355.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb5t1x.ent.gz | 290 KB | Display | PDB format |
| PDBx/mmJSON format | 5t1x.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/t1/5t1xftp://data.pdbj.org/pub/pdb/validation_reports/t1/5t1x | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  5t20C  1dlpS C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 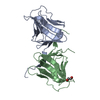
| ||||||||
| 2 | 
| ||||||||
| 3 | 
| ||||||||
| 4 | 
| ||||||||
| Unit cell |
|
-Components
-Protein , 2 types, 8 molecules ACEGBDFH
| #1: Protein | Mass: 12053.487 Da / Num. of mol.: 4 / Fragment: UNP residues 24-133 / Source method: isolated from a natural source / Source: (natural) Colocasia esculenta (taro) / References: UniProt: A5HMM7#2: Protein | Mass: 12309.715 Da / Num. of mol.: 4 / Source method: isolated from a natural source / Source: (natural) Colocasia esculenta (taro) / References: UniProt: Q39487*PLUS |
|---|
-Non-polymers , 4 types, 724 molecules 



| #3: Chemical | ChemComp-EDO /  Mass: 62.068 Da / Num. of mol.: 7 / Source method: obtained synthetically / Formula: C2H6O2 Mass: 62.068 Da / Num. of mol.: 7 / Source method: obtained synthetically / Formula: C2H6O2#4: Chemical | ChemComp-EPE / |  Mass: 238.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C8H18N2O4S / Comment: pH buffer*YM Mass: 238.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C8H18N2O4S / Comment: pH buffer*YM#5: Chemical | ChemComp-PGE / |  Mass: 150.173 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H14O4 Mass: 150.173 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H14O4#6: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 715 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Details
| Has protein modification | Y |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.49 Å3/Da / Density % sol: 50.54 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion / pH: 7 / Details: 25-35% Peg 3350, 0.2M Lithium Sulfate, 0.1M Hepes |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS / Beamline: 21-ID-G / Wavelength: 0.97857 Å |
| Detector | Type: RAYONIX MX-300 / Detector: CCD / Date: Dec 23, 2008 / Details: mirrors |
| Radiation | Monochromator: Diamond (111) / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.97857 Å / Relative weight: 1 |
| Reflection | Resolution: 1.7→50 Å / Num. obs: 104443 / % possible obs: 99.3 % / Redundancy: 4 % / Biso Wilson estimate: 18.41 Å2 / Rmerge(I) obs: 0.067 / Net I/σ(I): 11.4 |
| Reflection shell | Resolution: 1.7→1.75 Å / Redundancy: 3.9 % / Rmerge(I) obs: 0.381 / Rejects: 0 / % possible all: 98.1 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 1DLP Resolution: 1.7→17.14 Å / Cor.coef. Fo:Fc: 0.949 / Cor.coef. Fo:Fc free: 0.94 / Rfactor Rfree error: 0 / SU R Cruickshank DPI: 0.098 / Cross valid method: THROUGHOUT / σ(F): 0 / SU R Blow DPI: 0.1 / SU Rfree Blow DPI: 0.095 / SU Rfree Cruickshank DPI: 0.093
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 110.18 Å2 / Biso mean: 21.01 Å2 / Biso min: 4.74 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.21 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 1.7→17.14 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.7→1.74 Å / Total num. of bins used: 20
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
