: / Stability determinant / : / ParE toxin of type II toxin-antitoxin system, parDE / RelE-like / Toxin-antitoxin system, RelE/ParE toxin family / YaeB-like fold / Toxin-antitoxin system, RelE/ParE toxin domain superfamily / 2-Layer Sandwich / Alpha Beta Similarity search - Domain/homology
Type II toxin-antitoxin system RelE/ParE family toxin / : / Type II toxin-antitoxin system RelE/ParE family toxin / Stability determinant domain-containing protein Similarity search - Component
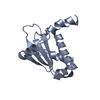
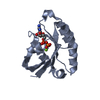
Journal: J Mol Biol / Year: 2016 Title: A unique hetero-hexadecameric architecture displayed by the Escherichia coli O157 PaaA2-ParE2 antitoxin-toxin complex. Authors: Yann G-J Sterckx / Thomas Jové / Alexander V Shkumatov / Abel Garcia-Pino / Lieselotte Geerts / Maia De Kerpel / Jurij Lah / Henri De Greve / Laurence Van Melderen / Remy Loris / Abstract: Many bacterial pathogens modulate their metabolic activity, virulence and pathogenicity through so-called "toxin-antitoxin" (TA) modules. The genome of the human pathogen Escherichia coli O157 ...Many bacterial pathogens modulate their metabolic activity, virulence and pathogenicity through so-called "toxin-antitoxin" (TA) modules. The genome of the human pathogen Escherichia coli O157 contains two three-component TA modules related to the known parDE module. Here, we show that the toxin EcParE2 maps in a branch of the RelE/ParE toxin superfamily that is distinct from the branches that contain verified gyrase and ribosome inhibitors. The structure of EcParE2 closely resembles that of Caulobacter crescentus ParE but shows a distinct pattern of conserved surface residues, in agreement with its apparent inability to interact with GyrA. The antitoxin EcPaaA2 is characterized by two α-helices (H1 and H2) that serve as molecular recognition elements to wrap itself around EcParE2. Both EcPaaA2 H1 and H2 are required to sustain a high-affinity interaction with EcParE2 and for the inhibition of EcParE2-mediated killing in vivo. Furthermore, evidence demonstrates that EcPaaA2 H2, but not H1, determines specificity for EcParE2. The initially formed EcPaaA2-EcParE2 heterodimer then assembles into a hetero-hexadecamer, which is stable in solution and is formed in a highly cooperative manner. Together these findings provide novel data on quaternary structure, TA interactions and activity of a hitherto poorly characterized family of TA modules.
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information
 X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation

 Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads












 PDBj
PDBj Assembly
Assembly



 Mass: 96.063 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: SO4
Mass: 96.063 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: SO4 Mass: 18.015 Da / Num. of mol.: 98 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 98 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation / Beamline: PROXIMA 1 / Wavelength: 0.97911 Å
/ Beamline: PROXIMA 1 / Wavelength: 0.97911 Å Processing
Processing