 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-4hdo: Crystal structure of the binary Complex of KRIT1 bound to the Rap... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4hdo | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of the binary Complex of KRIT1 bound to the Rap1 GTPase | ||||||
 Components Components |
| ||||||
 Keywords Keywords | SIGNALING PROTEIN / RA binding motif / PTB fold / GTPase / GTP / Rap effector / Rap1 / HEG1 / Cell-cell junctions / nucleus | ||||||
| Function / homology |  Function and homology information Function and homology informationRap protein signal transduction / GTPase regulator activity / regulation of cell junction assembly / endothelium development / positive regulation of integrin activation / modification of postsynaptic structure / negative regulation of calcium ion-dependent exocytosis / calcium-ion regulated exocytosis / negative regulation of synaptic vesicle exocytosis / integrin activation ...Rap protein signal transduction / GTPase regulator activity / regulation of cell junction assembly / endothelium development / positive regulation of integrin activation / modification of postsynaptic structure / negative regulation of calcium ion-dependent exocytosis / calcium-ion regulated exocytosis / negative regulation of synaptic vesicle exocytosis / integrin activation / Rap1 signalling / MET activates RAP1 and RAC1 / establishment of endothelial barrier / negative regulation of endothelial cell migration / azurophil granule membrane / p130Cas linkage to MAPK signaling for integrins / small GTPase-mediated signal transduction / regulation of establishment of cell polarity / negative regulation of endothelial cell proliferation / GRB2:SOS provides linkage to MAPK signaling for Integrins / regulation of angiogenesis / negative regulation of endothelial cell apoptotic process / Integrin signaling / phosphatidylinositol-4,5-bisphosphate binding / establishment of localization in cell / lipid droplet / Gene and protein expression by JAK-STAT signaling after Interleukin-12 stimulation / cell redox homeostasis / negative regulation of angiogenesis / cellular response to cAMP / small monomeric GTPase / Signaling by high-kinase activity BRAF mutants / MAP2K and MAPK activation / Signaling by RAF1 mutants / cell-cell junction / Signaling by moderate kinase activity BRAF mutants / Paradoxical activation of RAF signaling by kinase inactive BRAF / Signaling downstream of RAS mutants / Signaling by BRAF and RAF1 fusions / GDP binding / angiogenesis / G protein activity / microtubule binding / cytoskeleton / positive regulation of ERK1 and ERK2 cascade / cell population proliferation / GTPase activity / Neutrophil degranulation / GTP binding / protein-containing complex binding / glutamatergic synapse / protein-containing complex / : / extracellular exosome / membrane / plasma membrane / cytoplasm / cytosol Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.67 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.67 Å | ||||||
 Authors Authors | Gingras, A.R. | ||||||
 Citation Citation | Journal: J.Biol.Chem. / Year: 2013 Title: The Structure of the Ternary Complex of Krev Interaction Trapped 1 (KRIT1) Bound to Both the Rap1 GTPase and the Heart of Glass (HEG1) Cytoplasmic Tail. Authors: Gingras, A.R. / Puzon-McLaughlin, W. / Ginsberg, M.H. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4hdo.cif.gz | 118.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4hdo.ent.gz | 88.1 KB | Display | PDB format |
| PDBx/mmJSON format | 4hdo.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/hd/4hdoftp://data.pdbj.org/pub/pdb/validation_reports/hd/4hdo | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  4hdqC  1c1yS C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj













- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
-Protein , 2 types, 2 molecules AB
| #1: Protein | Mass: 37372.348 Da / Num. of mol.: 1 / Fragment: FERM domain (UNP residues 417-736) Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: KRIT1, CCM1 / Plasmid: pLEICS-07 / Production host:  |
|---|---|
| #2: Protein | Mass: 19020.508 Da / Num. of mol.: 1 / Fragment: UNP residues 1-167 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: RAP1B, OK/SW-cl.11 / Plasmid: pTAC / Production host: |
-Non-polymers , 4 types, 271 molecules 

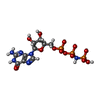

| #3: Chemical | ChemComp-GOL /  Mass: 92.094 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 92.094 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O3 |
|---|---|
| #4: Chemical | ChemComp-MG /  Mass: 24.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mg Mass: 24.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mg |
| #5: Chemical | ChemComp-GNP /  Mass: 522.196 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H17N6O13P3 Mass: 522.196 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H17N6O13P3Comment: GppNHp, GMPPNP, energy-carrying molecule analogue*YM |
| #6: Water | ChemComp-HOH / Mass: 18.015 Da / Num. of mol.: 268 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.35 Å3/Da / Density % sol: 47.58 % |
|---|---|
| Crystal grow | Temperature: 278 K / Method: vapor diffusion, sitting drop / pH: 8.5 Details: 15% PEG2000 MME, 100 mM Tris, 100 mM potassium chloride, pH 8.5, VAPOR DIFFUSION, SITTING DROP, temperature 278K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: Diamond  / Beamline: I02 / Wavelength: 0.9795 Å / Beamline: I02 / Wavelength: 0.9795 Å |
| Detector | Type: ADSC QUANTUM 315 / Detector: CCD / Date: Jul 23, 2010 |
| Radiation | Monochromator: double crystal Si(111) / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9795 Å / Relative weight: 1 |
| Reflection | Resolution: 1.67→50 Å / Num. all: 57913 / Num. obs: 57913 / % possible obs: 96 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Redundancy: 3.6 % / Rmerge(I) obs: 0.046 / Net I/σ(I): 31.27 |
| Reflection shell | Resolution: 1.67→1.77 Å / Redundancy: 2.8 % / Rmerge(I) obs: 0.35 / Mean I/σ(I) obs: 4.37 / Num. unique all: 21549 / % possible all: 80 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1C1Y Resolution: 1.67→32.43 Å / Cor.coef. Fo:Fc: 0.947 / Cor.coef. Fo:Fc free: 0.936 / SU B: 2.1 / SU ML: 0.072 / Cross valid method: THROUGHOUT / ESU R: 0.12 / ESU R Free: 0.11 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS U VALUES : REFINED INDIVIDUALLY
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 19.771 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.67→32.43 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.67→1.715 Å / Total num. of bins used: 20
|
