 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-4etp: C-terminal motor and motor homology domain of Kar3Vik1 fused to a... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4etp | ||||||
|---|---|---|---|---|---|---|---|
| Title | C-terminal motor and motor homology domain of Kar3Vik1 fused to a synthetic heterodimeric coiled coil | ||||||
 Components Components |
| ||||||
 Keywords Keywords | MOTOR PROTEIN / Kinesin Motor Protein / Kinesin Motor Homology Domain / karyogamy / mitosis / microtubules / internal Vik1 crosslink with N / N-Ethylenebis(iodoacetamide) / Nucleus | ||||||
| Function / homology |  Function and homology information Function and homology informationminus-end-directed kinesin ATPase / nuclear migration involved in conjugation with cellular fusion / protein transport along microtubule to mitotic spindle pole body / karyogamy involved in conjugation with cellular fusion / mitotic spindle pole body / mitotic spindle midzone assembly / cytoskeletal adaptor activity / microtubule nucleation / spindle pole body / minus-end-directed microtubule motor activity ...minus-end-directed kinesin ATPase / nuclear migration involved in conjugation with cellular fusion / protein transport along microtubule to mitotic spindle pole body / karyogamy involved in conjugation with cellular fusion / mitotic spindle pole body / mitotic spindle midzone assembly / cytoskeletal adaptor activity / microtubule nucleation / spindle pole body / minus-end-directed microtubule motor activity / mitotic sister chromatid cohesion / kinesin complex / microtubule-based process / cytoplasmic microtubule / ERAD pathway / regulation of mitotic spindle organization / meiotic cell cycle / spindle pole / mitotic cell cycle / microtubule cytoskeleton / chromosome / microtubule binding / microtubule / cell division / ATP hydrolysis activity / ATP binding / nucleus Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.3 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.3 Å | ||||||
 Authors Authors | Rank, K.C. / Chen, C.J. / Cope, J. / Porche, K. / Hoenger, A. / Gilbert, S.P. / Rayment, I. | ||||||
 Citation Citation | Journal: J Cell Biol / Year: 2012 Title: Kar3Vik1, a member of the kinesin-14 superfamily, shows a novel kinesin microtubule binding pattern. Authors: Katherine C Rank / Chun Ju Chen / Julia Cope / Ken Porche / Andreas Hoenger / Susan P Gilbert / Ivan Rayment /  Abstract: Kinesin-14 motors generate microtubule minus-end-directed force used in mitosis and meiosis. These motors are dimeric and operate with a nonprocessive powerstroke mechanism, but the role of the ...Kinesin-14 motors generate microtubule minus-end-directed force used in mitosis and meiosis. These motors are dimeric and operate with a nonprocessive powerstroke mechanism, but the role of the second head in motility has been unclear. In Saccharomyces cerevisiae, the Kinesin-14 Kar3 forms a heterodimer with either Vik1 or Cik1. Vik1 contains a motor homology domain that retains microtubule binding properties but lacks a nucleotide binding site. In this case, both heads are implicated in motility. Here, we show through structural determination of a C-terminal heterodimeric Kar3Vik1, electron microscopy, equilibrium binding, and motility that at the start of the cycle, Kar3Vik1 binds to or occludes two αβ-tubulin subunits on adjacent protofilaments. The cycle begins as Vik1 collides with the microtubule followed by Kar3 microtubule association and ADP release, thereby destabilizing the Vik1-microtubule interaction and positioning the motor for the start of the powerstroke. The results indicate that head-head communication is mediated through the adjoining coiled coil. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4etp.cif.gz | 296.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4etp.ent.gz | 237.4 KB | Display | PDB format |
| PDBx/mmJSON format | 4etp.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/et/4etpftp://data.pdbj.org/pub/pdb/validation_reports/et/4etp | HTTPS FTP |
|---|
-Related structure data
















-Links
 PDBj
PDBj




- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
-Protein , 2 types, 2 molecules AB
| #1: Protein | Mass: 45178.941 Da / Num. of mol.: 1 / Fragment: Kar3 (unp residues 352-729) / Mutation: C391L, C469A, C517A, C566V, C655V) Source method: isolated from a genetically manipulated source Source: (gene. exp.) Strain: ATCC 204508 / S288c / Gene: KAR3, kar3p, P9659.16, YPR141C / Plasmid: pET24d / Production host:  |
|---|---|
| #2: Protein | Mass: 38478.742 Da / Num. of mol.: 1 / Fragment: Vik1 (unp residues 341-647) / Mutation: C377V, C436A, C536A, C596A, C640A, E355C, K423C) Source method: isolated from a genetically manipulated source Source: (gene. exp.) Strain: ATCC 204508 / S288c / Gene: VIK1, Vik1p, YPL253C / Plasmid: pKLD37 / Production host: |
-Non-polymers , 5 types, 255 molecules 


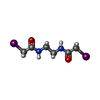

| #3: Chemical | ChemComp-ADP /  Mass: 427.201 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H15N5O10P2 / Comment: ADP, energy-carrying molecule*YM Mass: 427.201 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H15N5O10P2 / Comment: ADP, energy-carrying molecule*YM | ||||
|---|---|---|---|---|---|
| #4: Chemical | ChemComp-MG /  Mass: 24.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mg Mass: 24.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mg | ||||
| #5: Chemical | ChemComp-GOL /  Mass: 92.094 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 92.094 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: C3H8O3#6: Chemical | ChemComp-EBC / |  Mass: 395.965 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H10I2N2O2 Mass: 395.965 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H10I2N2O2#7: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 246 / Source method: isolated from a natural source / Formula: H2O |
-Details
| Has protein modification | Y |
|---|---|
| Nonpolymer details | THE STARTING MATERIAL USED FOR CROSS LINKING CONTAINED A IODINE ATOM THAT HAS LEAVED UPON REACTION ...THE STARTING MATERIAL USED FOR CROSS LINKING CONTAINED A IODINE ATOM THAT HAS LEAVED UPON REACTION WITH CYS. THE FINAL PRODUCT CORRESPOND |
| Sequence details | NATIVE KAR3VIK1 CONTAINS EXTENSIVE BUT WEAK COILED-COIL. FOR CRYSTALLIZATION, ALL BUT THE LAST C- ...NATIVE KAR3VIK1 CONTAINS EXTENSIVE BUT WEAK COILED-COIL. FOR CRYSTALLIZ |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.86 Å3/Da / Density % sol: 57.04 % |
|---|---|
| Crystal grow | Temperature: 277 K / Method: evaporation / pH: 7.5 Details: 16% monomethyl polyethylene glycol 2000, 100 mM Hepes, 50 mM sodium citrate, Batch, pH 7.5, EVAPORATION, temperature 277K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS / Beamline: 19-ID / Wavelength: 0.979206 Å |
| Detector | Type: ADSC QUANTUM 315 / Detector: CCD / Date: Dec 2, 2010 |
| Radiation | Monochromator: Rosenbaum-Rock high-resolution double-crystal monochromator. LN2 cooled first crystal, sagittal focusing 2nd crystal Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.979206 Å / Relative weight: 1 |
| Reflection | Resolution: 2.3→25 Å / Num. all: 43354 / Num. obs: 43179 / % possible obs: 99.9 % / Observed criterion σ(F): 0 / Observed criterion σ(I): -3 / Redundancy: 8.6 % / Rmerge(I) obs: 0.051 / Rsym value: 0.069 / Net I/σ(I): 19.7 |
| Reflection shell | Resolution: 2.3→2.34 Å / Redundancy: 6.2 % / Rmerge(I) obs: 0.601 / Mean I/σ(I) obs: 3.2 / Num. unique all: 2701 / Rsym value: 0.593 / % possible all: 99.8 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: pdb entry 3KAR and 2O0A Resolution: 2.3→24.97 Å / Cor.coef. Fo:Fc: 0.95 / Cor.coef. Fo:Fc free: 0.936 / SU B: 10.825 / SU ML: 0.142 / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R: 0.274 / ESU R Free: 0.215 / Stereochemistry target values: MAXIMUM LIKELIHOOD / Details: HYDROGENS HAVE BEEN USED IF PRESENT IN THE INPUT
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1 Å / Solvent model: MASK | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 69.055 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.3→24.97 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.301→2.361 Å / Total num. of bins used: 20
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
