 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-3f8i: Mouse UHRF1 SRA domain bound with hemi-methylated CpG, crystal st... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3f8i | ||||||
|---|---|---|---|---|---|---|---|
| Title | Mouse UHRF1 SRA domain bound with hemi-methylated CpG, crystal structure in space group P21 | ||||||
 Components Components |
| ||||||
 Keywords Keywords | LIGASE/DNA / UHRF1 / base flipping / 5-methylcytosine / CpG methylation / Cell cycle / Developmental protein / DNA damage / DNA repair / DNA-binding / Ligase / Metal-binding / Nucleus / Phosphoprotein / Transcription / Transcription regulation / Ubl conjugation / Ubl conjugation pathway / Zinc / Zinc-finger / LIGASE-DNA COMPLEX | ||||||
| Function / homology |  Function and homology information Function and homology informationhistone H3K18 ubiquitin ligase activity / histone H3K14 ubiquitin ligase activity / histone H3 ubiquitin ligase activity / histone H3K23 ubiquitin ligase activity / histone H3 reader activity / chromosomal DNA methylation maintenance following DNA replication / hemi-methylated DNA-binding / regulation of epithelial cell proliferation / methyl-CpG binding / histone H3K9me2/3 reader activity ...histone H3K18 ubiquitin ligase activity / histone H3K14 ubiquitin ligase activity / histone H3 ubiquitin ligase activity / histone H3K23 ubiquitin ligase activity / histone H3 reader activity / chromosomal DNA methylation maintenance following DNA replication / hemi-methylated DNA-binding / regulation of epithelial cell proliferation / methyl-CpG binding / histone H3K9me2/3 reader activity / negative regulation of gene expression via chromosomal CpG island methylation / positive regulation of protein metabolic process / mitotic spindle assembly / protein autoubiquitination / cis-regulatory region sequence-specific DNA binding / heterochromatin / replication fork / euchromatin / RING-type E3 ubiquitin transferase / nuclear matrix / ubiquitin-protein transferase activity / ubiquitin protein ligase activity / heterochromatin formation / histone binding / nucleic acid binding / ubiquitin-dependent protein catabolic process / protein ubiquitination / DNA repair / chromatin / negative regulation of transcription by RNA polymerase II / positive regulation of transcription by RNA polymerase II / zinc ion binding / nucleoplasm / identical protein binding / nucleus / cytosol Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.29 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.29 Å | ||||||
 Authors Authors | Hashimoto, H. / Horton, J.R. / Zhang, X. / Cheng, X. | ||||||
 Citation Citation | Journal: Epigenetics / Year: 2009 Title: UHRF1, a modular multi-domain protein, regulates replication-coupled crosstalk between DNA methylation and histone modifications. Authors: Hashimoto, H. / Horton, J.R. / Zhang, X. / Cheng, X. #1: Journal: Nature / Year: 2008Title: The SRA domain of UHRF1 flips 5-methylcytosine out of the DNA helix. Authors: Hashimoto, H. / Horton, J.R. / Zhang, X. / Bostick, M. / Jacobsen, S.E. / Cheng, X. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3f8i.cif.gz | 124.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3f8i.ent.gz | 91.9 KB | Display | PDB format |
| PDBx/mmJSON format | 3f8i.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/f8/3f8iftp://data.pdbj.org/pub/pdb/validation_reports/f8/3f8i | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  3f8jC  3fdeC  2zo0S S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |












-Links
 PDBj
PDBj









































- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 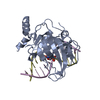
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
| ||||||||
| Details | Asymmetric unit contains two SRA-DNA complexes. |
-Components
| #1: Protein | Mass: 23915.711 Da / Num. of mol.: 2 / Fragment: YDG domain: UNP residues 419-628 Source method: isolated from a genetically manipulated source Details: hexahistidine-SUMO-tagged construct / Source: (gene. exp.)  #2: DNA chain | Mass: 3637.395 Da / Num. of mol.: 2 / Source method: obtained synthetically / Details: Synthetic DNA oligo #3: DNA chain | Mass: 3703.416 Da / Num. of mol.: 2 / Source method: obtained synthetically / Details: Synthetic DNA oligo #4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 135 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 135 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.81 Å3/Da / Density % sol: 56.15 % | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 277 K / pH: 7 Details: 20% PEG 3350, 0.4M NaCl, pH 7.0, VAPOR DIFFUSION, temperature 277K | ||||||||||||||||||||
| Components of the solutions |
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS  / Beamline: 22-ID / Wavelength: 1 / Beamline: 22-ID / Wavelength: 1 |
| Detector | Type: MARMOSAIC 300 mm CCD / Detector: CCD / Date: Apr 20, 2008 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 |
| Reflection | Resolution: 2.29→34.53 Å / Num. obs: 30041 / % possible obs: 97.5 % / Observed criterion σ(I): -3 / Redundancy: 3.4 % / Biso Wilson estimate: 16.7 Å2 / Rmerge(I) obs: 0.102 / Net I/σ(I): 10.5 |
| Reflection shell | Resolution: 2.29→2.37 Å / Redundancy: 2.9 % / Rmerge(I) obs: 0.352 / Mean I/σ(I) obs: 2.3 / % possible all: 90.5 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 2ZO0 Resolution: 2.29→34.53 Å / Rfactor Rfree error: 0.007 / Data cutoff high absF: 622448.07 / Data cutoff low absF: 0 / Cross valid method: THROUGHOUT / σ(F): 0 / Stereochemistry target values: ENGH & HUBER
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 32.0138 Å2 / ksol: 0.4 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 35.1 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.29→34.53 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.29→2.37 Å / Rfactor Rfree error: 0.042 / Total num. of bins used: 10
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
|
