 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2x9d | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Tet repressor (class D) in complex with iso-7-chlortetracycline | |||||||||
 Components Components | TETRACYCLINE REPRESSOR PROTEIN CLASS D | |||||||||
 Keywords Keywords | TRANSCRIPTION / GENE REGULATION / HTH-MOTIF / DNA-BINDING PROTEIN / ANTIBIOTIC RESISTANCE | |||||||||
| Function / homology |  Function and homology information Function and homology informationtranscription cis-regulatory region binding / DNA-binding transcription factor activity / response to antibiotic / negative regulation of DNA-templated transcription / metal ion binding Similarity search - Function | |||||||||
| Biological species |  | |||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.34 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.34 Å | |||||||||
 Authors Authors | Volkers, G. / Hinrichs, W. | |||||||||
 Citation Citation | Journal: J.Med.Chem. / Year: 2011 Title: Recognition of Drug Degradation Products by Target Proteins: Isotetracycline Binding to Tet Repressor. Authors: Volkers, G. / Petruschka, L. / Hinrichs, W. #1: Journal: Science / Year: 1994Title: Structure of the Tet Repressor-Tetracycline Complex and Regulation of Antibiotic Resistance. Authors: Hinrichs, W. / Kisker, C. / Duvel, M. / Muller, A. / Tovar, K. / Hillen, W. / Saenger, W. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2x9d.cif.gz | 94.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2x9d.ent.gz | 72.7 KB | Display | PDB format |
| PDBx/mmJSON format | 2x9d.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/x9/2x9dftp://data.pdbj.org/pub/pdb/validation_reports/x9/2x9d | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  2x6oC  2tctS C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |





























-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 23288.334 Da / Num. of mol.: 1 / Fragment: RESIDUES 2-208 / Mutation: YES Source method: isolated from a genetically manipulated source Details: C-TERMINUS REMOVED FOR BETTER CRYSTALLIZATION / Source: (gene. exp.) | ||||||
|---|---|---|---|---|---|---|---|
| #2: Chemical | ChemComp-ITC / 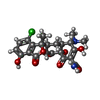  Mass: 478.880 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C22H23ClN2O8 Mass: 478.880 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C22H23ClN2O8 | ||||||
| #3: Chemical | ChemComp-CL /   Mass: 35.453 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Cl Mass: 35.453 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Cl#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 64 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 64 / Source method: isolated from a natural source / Formula: H2OCompound details | ENGINEERED | Sequence details | A2S CLONING ARTEFACT | |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.27 Å3/Da / Density % sol: 45.3 % / Description: NONE |
|---|---|
| Crystal grow | Method: vapor diffusion, hanging drop / pH: 8 Details: 0.002ML PROTEIN/COMPLEX (0.050 ML TETR(D) (C = 17.3 MG/ML)50 ML 2MM ISO-CLTC) PLUS ML BUFFER (50 MM TRIS PH 8, 1.1 M (NH4)2SO4, 100 MM NACL, 10 MM MGCL2) HANGING DROP AGAINST 0.5 ML BUFFER |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: EMBL/DESY, HAMBURG  / Beamline: X13 / Wavelength: 0.979 / Beamline: X13 / Wavelength: 0.979 |
| Detector | Type: MARRESEARCH SX-165 / Detector: CCD / Date: Mar 26, 2004 / Details: MIRRORS |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.979 Å / Relative weight: 1 |
| Reflection | Resolution: 2.34→63.7 Å / Num. obs: 9121 / % possible obs: 98 % / Observed criterion σ(I): 0 / Redundancy: 4.48 % / Biso Wilson estimate: 45.2 Å2 / Rmerge(I) obs: 0.07 / Net I/σ(I): 9.7 |
| Reflection shell | Resolution: 2.34→2.39 Å / Redundancy: 3.44 % / Rmerge(I) obs: 0.64 / Mean I/σ(I) obs: 1.78 / % possible all: 76 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 2TCT Resolution: 2.34→63.76 Å / Cor.coef. Fo:Fc: 0.923 / Cor.coef. Fo:Fc free: 0.904 / SU B: 28.675 / SU ML: 0.36 / Cross valid method: THROUGHOUT / ESU R: 0.576 / ESU R Free: 0.306 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS. RESIDUES 153-164 ARE DISORDERED. THE TRACING OF THE TURN RESIDUES. LEU 101 TO PRO 105 IS AMBIGUOUS BECAUSE OF WEAK ELECTRON DENSITY.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: BABINET MODEL WITH MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 48.08 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.34→63.76 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|