Entry Database : PDB / ID : 2x2dTitle acetyl-CypA:HIV-1 N-term capsid domain complex CAPSID PROTEIN P24 PEPTIDYL-PROLYL CIS-TRANS ISOMERASE A Keywords / / / / / Function / homology Function Domain/homology Component
/ / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / Biological species HOMO SAPIENS (human)Method / / OTHER / Resolution : 1.95 Å Authors Lammers, M. / Neumann, H. / Chin, J.W. / James, L.C. Journal : Nat.Chem.Biol. / Year : 2010Title : Acetylation Regulates Cyclophilin a Catalysis, Immunosuppression and HIV IsomerisationAuthors : Lammers, M. / Neumann, H. / Chin, J.W. / James, L.C. History Deposition Jan 12, 2010 Deposition site / Processing site Revision 1.0 Mar 23, 2010 Provider / Type Revision 1.1 Jul 13, 2011 Group / Version format complianceRevision 1.2 Oct 26, 2011 Group / Derived calculationsRevision 1.3 Jan 24, 2018 Group / Category Item _entity_src_gen.pdbx_host_org_ncbi_taxonomy_id / _entity_src_gen.pdbx_host_org_scientific_name ... _entity_src_gen.pdbx_host_org_ncbi_taxonomy_id / _entity_src_gen.pdbx_host_org_scientific_name / _entity_src_gen.pdbx_host_org_strain / _entity_src_gen.pdbx_host_org_variant Revision 1.4 Nov 13, 2024 Group Data collection / Database references ... Data collection / Database references / Derived calculations / Other / Structure summary Category chem_comp_atom / chem_comp_bond ... chem_comp_atom / chem_comp_bond / database_2 / pdbx_database_status / pdbx_entry_details / pdbx_modification_feature / struct_conn Item _database_2.pdbx_DOI / _database_2.pdbx_database_accession ... _database_2.pdbx_DOI / _database_2.pdbx_database_accession / _pdbx_database_status.status_code_sf / _struct_conn.pdbx_leaving_atom_flag
Show all Show less
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information HOMO SAPIENS (human)
HOMO SAPIENS (human)
 HUMAN IMMUNODEFICIENCY VIRUS 1
HUMAN IMMUNODEFICIENCY VIRUS 1 X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads










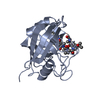
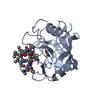























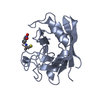

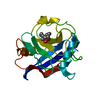





















 PDBj
PDBj












 Assembly
Assembly



 Mass: 18.015 Da / Num. of mol.: 208 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 208 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation / Beamline: ID29 / Wavelength: 0.7
/ Beamline: ID29 / Wavelength: 0.7  Processing
Processing