 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
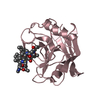
| Entry | Database: PDB / ID: 3cyh | ||||||
|---|---|---|---|---|---|---|---|
| Title | CYCLOPHILIN A COMPLEXED WITH DIPEPTIDE SER-PRO | ||||||
 Components Components | CYCLOPHILIN A | ||||||
 Keywords Keywords | ISOMERASE / CYCLOPHILIN / COMPLEX / BINDING PROTEIN FOR CYCLOSPORIN A / (ISOMERASE-DIPEPTIDE) COMPLEX | ||||||
| Function / homology |  Function and homology information Function and homology information: / negative regulation of protein K48-linked ubiquitination / regulation of apoptotic signaling pathway / cell adhesion molecule production / lipid droplet organization / negative regulation of viral life cycle / heparan sulfate binding / regulation of viral genome replication / virion binding / leukocyte chemotaxis ...: / negative regulation of protein K48-linked ubiquitination / regulation of apoptotic signaling pathway / cell adhesion molecule production / lipid droplet organization / negative regulation of viral life cycle / heparan sulfate binding / regulation of viral genome replication / virion binding / leukocyte chemotaxis / negative regulation of stress-activated MAPK cascade / activation of protein kinase B activity / endothelial cell activation / Basigin interactions / protein peptidyl-prolyl isomerization / cyclosporin A binding / Minus-strand DNA synthesis / Plus-strand DNA synthesis / Uncoating of the HIV Virion / Early Phase of HIV Life Cycle / Integration of provirus / APOBEC3G mediated resistance to HIV-1 infection / negative regulation of protein phosphorylation / intracellular membrane-bounded organelle / viral release from host cell / Binding and entry of HIV virion / Calcineurin activates NFAT / negative regulation of oxidative stress-induced intrinsic apoptotic signaling pathway / negative regulation of protein kinase activity / positive regulation of viral genome replication / neutrophil chemotaxis / Gene and protein expression by JAK-STAT signaling after Interleukin-12 stimulation / : / peptidylprolyl isomerase / positive regulation of protein secretion / peptidyl-prolyl cis-trans isomerase activity / Assembly Of The HIV Virion / platelet activation / Budding and maturation of HIV virion / positive regulation of protein phosphorylation / platelet aggregation / integrin binding / neuron differentiation / SARS-CoV-1 activates/modulates innate immune responses / : / Platelet degranulation / protein folding / cellular response to oxidative stress / secretory granule lumen / vesicle / ficolin-1-rich granule lumen / positive regulation of MAPK cascade / focal adhesion / apoptotic process / Neutrophil degranulation / protein-containing complex / : / RNA binding / extracellular exosome / extracellular region / membrane / nucleus / cytoplasm / cytosol Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / Resolution: 1.9 Å X-RAY DIFFRACTION / Resolution: 1.9 Å | ||||||
 Authors Authors | Zhao, Y. / Ke, H. | ||||||
 Citation Citation | Journal: Biochemistry / Year: 1996 Title: Mechanistic implication of crystal structures of the cyclophilin-dipeptide complexes. Authors: Zhao, Y. / Ke, H. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3cyh.cif.gz | 51.9 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3cyh.ent.gz | 37.4 KB | Display | PDB format |
| PDBx/mmJSON format | 3cyh.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/cy/3cyhftp://data.pdbj.org/pub/pdb/validation_reports/cy/3cyh | HTTPS FTP |
|---|
-Related structure data












-Links
 PDBj
PDBj












- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 17905.307 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Cell line: XA90 / Gene: CYCLOPHILIN / Plasmid: PHN1+ / Gene (production host): CYCLOPHILIN / Production host:  References: UniProt: P05092, UniProt: P62937*PLUS, peptidylprolyl isomerase |
|---|---|
| #2: Chemical | ChemComp-SER /   Type: L-peptide linking / Mass: 105.093 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H7NO3 Type: L-peptide linking / Mass: 105.093 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H7NO3 |
| #3: Chemical | ChemComp-PRO / 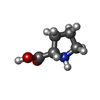  Type: L-peptide linking / Mass: 115.130 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C5H9NO2 Type: L-peptide linking / Mass: 115.130 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C5H9NO2 |
| #4: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 54 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 54 / Source method: isolated from a natural source / Formula: H2O |
| Nonpolymer details | THE RESIDUES 201 AND 202 REPRESENT A DIPEPTIDE BOUND TO CYCLOPHILI |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.67 Å3/Da / Density % sol: 54 % | ||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | *PLUS Temperature: 4 ℃ / pH: 8.5 / Method: microdialysis | ||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction source | Wavelength: 1.5418 |
|---|---|
| Detector | Type: RIGAKU RAXIS IIC / Detector: IMAGE PLATE |
| Radiation | Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Highest resolution: 1.9 Å / Num. obs: 14311 / % possible obs: 90 % / Redundancy: 4 % / Rmerge(I) obs: 0.056 |
| Reflection | *PLUS Num. measured all: 56836 |
| Reflection shell | *PLUS Highest resolution: 1.9 Å / Lowest resolution: 2 Å / % possible obs: 74.4 % |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Resolution: 1.9→8 Å / σ(F): 2 /
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 19.5 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.9→8 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: X-PLOR / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Lowest resolution: 8 Å / Rfactor obs: 0.186 / Rfactor Rfree: 0.231 / Rfactor Rwork: 0.186 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS |
