 Movie
Movie Controller
Controller

[English] 日本語
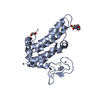
 Yorodumi
Yorodumi- PDB-2wwk: Crystal structure of the Titin M10-Obscurin like 1 Ig F17R mutant... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2wwk | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of the Titin M10-Obscurin like 1 Ig F17R mutant complex | ||||||
 Components Components |
| ||||||
 Keywords Keywords | TRANSFERASE/STRUCTURAL PROTEIN / TRANSFERASE-STRUCTURAL PROTEIN COMPLEX / SARCOMERE / IMMUNOGLOBULIN DOMAIN / LIMB-GIRDLE MUSCULAR DYSTROPHY / KELCH REPEAT / CARDIOMYOPATHY | ||||||
| Function / homology |  Function and homology information Function and homology information3M complex / sarcomerogenesis / titin-telethonin complex / structural molecule activity conferring elasticity / skeletal muscle myosin thick filament assembly / telethonin binding / protein localization to Golgi apparatus / detection of muscle stretch / muscle alpha-actinin binding / : ...3M complex / sarcomerogenesis / titin-telethonin complex / structural molecule activity conferring elasticity / skeletal muscle myosin thick filament assembly / telethonin binding / protein localization to Golgi apparatus / detection of muscle stretch / muscle alpha-actinin binding / : / cytoskeletal anchor activity / cardiac myofibril assembly / positive regulation of dendrite morphogenesis / cardiac muscle hypertrophy / mitotic chromosome condensation / cardiac muscle tissue morphogenesis / Striated Muscle Contraction / muscle filament sliding / protein kinase regulator activity / actinin binding / M band / I band / cardiac muscle cell development / structural constituent of muscle / sarcomere organization / regulation of mitotic nuclear division / Golgi organization / striated muscle thin filament / skeletal muscle thin filament assembly / intercalated disc / skeletal muscle contraction / striated muscle contraction / cardiac muscle contraction / cytoskeleton organization / muscle contraction / condensed nuclear chromosome / positive regulation of protein secretion / response to calcium ion / microtubule cytoskeleton organization / Z disc / actin filament binding / Platelet degranulation / Neddylation / protease binding / protein tyrosine kinase activity / calmodulin binding / non-specific serine/threonine protein kinase / protein kinase activity / protein serine kinase activity / protein serine/threonine kinase activity / calcium ion binding / centrosome / positive regulation of gene expression / protein kinase binding / perinuclear region of cytoplasm / enzyme binding / Golgi apparatus / protein homodimerization activity / extracellular exosome / extracellular region / ATP binding / identical protein binding / cytoplasm / cytosol Similarity search - Function | ||||||
| Biological species |  HOMO SAPIENS (human) HOMO SAPIENS (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.7 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.7 Å | ||||||
 Authors Authors | Pernigo, S. / Fukuzawa, A. / Gautel, M. / Steiner, R.A. | ||||||
 Citation Citation | Journal: Proc.Natl.Acad.Sci.USA / Year: 2010 Title: Structural Insight Into M-Band Assembly and Mechanics from the Titin-Obscurin-Like-1 Complex. Authors: Pernigo, S. / Fukuzawa, A. / Bertz, M. / Holt, M. / Rief, M. / Steiner, R.A. / Gautel, M. | ||||||
| History |
| ||||||
| Remark 700 | SHEET THE SHEET STRUCTURE OF THIS MOLECULE IS BIFURCATED. IN ORDER TO REPRESENT THIS FEATURE IN ... SHEET THE SHEET STRUCTURE OF THIS MOLECULE IS BIFURCATED. IN ORDER TO REPRESENT THIS FEATURE IN THE SHEET RECORDS BELOW, TWO SHEETS ARE DEFINED. |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2wwk.cif.gz | 61.4 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2wwk.ent.gz | 44.2 KB | Display | PDB format |
| PDBx/mmJSON format | 2wwk.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Summary document | 2wwk_validation.pdf.gz | 454.5 KB | Display | wwPDB validaton report |
|---|---|---|---|---|
| Full document | 2wwk_full_validation.pdf.gz | 457 KB | Display | |
| Data in XML | 2wwk_validation.xml.gz | 13.2 KB | Display | |
| Data in CIF | 2wwk_validation.cif.gz | 19.4 KB | Display | |
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ww/2wwkftp://data.pdbj.org/pub/pdb/validation_reports/ww/2wwk | HTTPS FTP |
-Related structure data
| Related structure data |  2wp3SC  2wwmC S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |


























-Links
 PDBj
PDBj




- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Components on special symmetry positions |
|
-Components
| #1: Protein | Mass: 11135.640 Da / Num. of mol.: 1 / Fragment: IG1, RESIDUES 1-106 / Mutation: YES Source method: isolated from a genetically manipulated source Source: (gene. exp.) HOMO SAPIENS (human) / Production host:  | ||
|---|---|---|---|
| #2: Protein | Mass: 10981.231 Da / Num. of mol.: 1 / Fragment: M10, RESIDUES 34252-34350 Source method: isolated from a genetically manipulated source Source: (gene. exp.) HOMO SAPIENS (human) / Production host: References: UniProt: Q8WZ42, non-specific serine/threonine protein kinase | ||
| #3: Chemical | ChemComp-GOL /   Mass: 92.094 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 92.094 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O3 | ||
| #4: Chemical | ChemComp-SO4 /   Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4 | ||
| #5: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 287 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 287 / Source method: isolated from a natural source / Formula: H2O | ||
| Compound details | ENGINEERED| Sequence details | INITIAL GSS ARE FROM THE EXPRESSION | |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.29 Å3/Da / Density % sol: 45.82 % / Description: NONE |
|---|---|
| Crystal grow | pH: 5.5 Details: 0.1 M BIS-TRIS PH 5.5, 0.2 M AMMONIUM SULPHATE, 25% PEG 3350 |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: Diamond  / Beamline: I04 / Wavelength: 0.9763 / Beamline: I04 / Wavelength: 0.9763 |
| Detector | Type: ADSC CCD / Detector: CCD / Date: Aug 12, 2009 / Details: MIRRORS |
| Radiation | Monochromator: SI (111) DOUBLE CRYSTAL / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9763 Å / Relative weight: 1 |
| Reflection | Resolution: 1.7→34.79 Å / Num. obs: 24975 / % possible obs: 98.7 % / Observed criterion σ(I): 0 / Redundancy: 7.8 % / Biso Wilson estimate: 20.95 Å2 / Rmerge(I) obs: 0.08 / Net I/σ(I): 20.6 |
| Reflection shell | Resolution: 1.7→1.79 Å / Redundancy: 8 % / Rmerge(I) obs: 0.31 / Mean I/σ(I) obs: 6 / % possible all: 99.7 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 2WP3 Resolution: 1.7→31.27 Å / Cor.coef. Fo:Fc: 0.921 / Cor.coef. Fo:Fc free: 0.891 / SU B: 5.219 / SU ML: 0.079 / TLS residual ADP flag: LIKELY RESIDUAL / Cross valid method: THROUGHOUT / ESU R: 0.126 / ESU R Free: 0.124 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS. U VALUES RESIDUAL ONLY. ATOM RECORD CONTAINS RESIDUAL B FACTORS ONLY
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 7.317 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.7→31.27 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|