 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1w8b | ||||||
|---|---|---|---|---|---|---|---|
| Title | Factor7 - 413 complex | ||||||
 Components Components | (BLOOD COAGULATION FACTOR VIIA) x 2 | ||||||
 Keywords Keywords | HYDROLASE / SERINE PROTEASE / COAGULATION / ENZYME COMPLEX | ||||||
| Function / homology |  Function and homology information Function and homology informationcoagulation factor VIIa / response to Thyroid stimulating hormone / response to astaxanthin / response to thyrotropin-releasing hormone / response to 2,3,7,8-tetrachlorodibenzodioxine / response to carbon dioxide / serine-type peptidase complex / response to genistein / response to vitamin K / response to thyroxine ...coagulation factor VIIa / response to Thyroid stimulating hormone / response to astaxanthin / response to thyrotropin-releasing hormone / response to 2,3,7,8-tetrachlorodibenzodioxine / response to carbon dioxide / serine-type peptidase complex / response to genistein / response to vitamin K / response to thyroxine / positive regulation of platelet-derived growth factor receptor signaling pathway / positive regulation of leukocyte chemotaxis / response to cholesterol / positive regulation of positive chemotaxis / response to growth hormone / : / animal organ regeneration / positive regulation of blood coagulation / positive regulation of TOR signaling / Transport of gamma-carboxylated protein precursors from the endoplasmic reticulum to the Golgi apparatus / Gamma-carboxylation of protein precursors / Removal of aminoterminal propeptides from gamma-carboxylated proteins / BMAL1:CLOCK,NPAS2 activates circadian expression / serine-type peptidase activity / circadian rhythm / protein processing / response to estrogen / Golgi lumen / blood coagulation / response to estradiol / extracellular matrix / vesicle / response to hypoxia / positive regulation of cell migration / endoplasmic reticulum lumen / serine-type endopeptidase activity / signaling receptor binding / calcium ion binding / : / extracellular region / plasma membrane Similarity search - Function | ||||||
| Biological species |  HOMO SAPIENS (human) HOMO SAPIENS (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 3 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 3 Å | ||||||
 Authors Authors | Ackermann, J. / Alig, L. / Banner, D.W. / Boehm, H.-J. / Groebke-Zbinden, K. / Hilpert, K. / Lave, T. / Kuehne, H. / Obst-Sander, U. / Riederer, M.A. ...Ackermann, J. / Alig, L. / Banner, D.W. / Boehm, H.-J. / Groebke-Zbinden, K. / Hilpert, K. / Lave, T. / Kuehne, H. / Obst-Sander, U. / Riederer, M.A. / Stahl, M. / Tschopp, T.B. / Weber, L. / Wessel, H.P. | ||||||
 Citation Citation | Journal: Bioorg.Med.Chem.Lett. / Year: 2005 Title: Selective and Orally Bioavailable Phenylglycine Tissue Factor/Factor Viia Inhibitors Authors: Groebke-Zbinden, K. / Obst-Sander, U. / Hilpert, K. / Kuehne, H. / Banner, D.W. / Boehm, H.-J. / Stahl, M. / Ackermann, J. / Alig, L. / Weber, L. / Wessel, H.P. / Riederer, M.A. / Tschopp, T.B. / Lave, T. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1w8b.cif.gz | 76.6 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1w8b.ent.gz | 56.2 KB | Display | PDB format |
| PDBx/mmJSON format | 1w8b.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/w8/1w8bftp://data.pdbj.org/pub/pdb/validation_reports/w8/1w8b | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1w7xC  1w0yS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |


























-Links
 PDBj
PDBj





- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 28103.256 Da / Num. of mol.: 1 / Fragment: FACTOR VII HEAVY CHAIN, RESIDUES 213-466 Source method: isolated from a genetically manipulated source Source: (gene. exp.) HOMO SAPIENS (human) / Production host:  |
|---|---|
| #2: Protein | Mass: 6272.113 Da / Num. of mol.: 1 / Fragment: FACTOR VII LIGHT CHAIN, RESIDUES 148-204 Source method: isolated from a genetically manipulated source Source: (gene. exp.) HOMO SAPIENS (human) / Production host: |
| #3: Chemical | ChemComp-CA /   Mass: 40.078 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Ca Mass: 40.078 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Ca |
| #4: Chemical | ChemComp-413 / (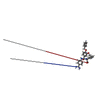  Mass: 538.594 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C31H30N4O5 Mass: 538.594 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C31H30N4O5 |
| #5: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 59 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 59 / Source method: isolated from a natural source / Formula: H2O |
| Has protein modification | Y |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.6 Å3/Da / Density % sol: 65 % Description: ONLY 27DEGREES OF DATA WERE COLLECTED, SO COMPLETENESS IS VERY POOR. ALSO THERE WAS SEVERE ICING. |
|---|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: BM1A / Wavelength: 0.873 / Beamline: BM1A / Wavelength: 0.873 |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE / Date: Apr 24, 1998 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.873 Å / Relative weight: 1 |
| Reflection | Resolution: 2.82→20 Å / Num. obs: 23089 / % possible obs: 75 % / Observed criterion σ(I): 0 / Redundancy: 2.3 % / Rmerge(I) obs: 0.16 / Net I/σ(I): 4.3 |
| Reflection shell | Resolution: 2.82→2.92 Å / % possible all: 62 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1W0Y Resolution: 3→72.36 Å / Cor.coef. Fo:Fc: 0.913 / Cor.coef. Fo:Fc free: 0.843 / SU B: 14.796 / SU ML: 0.278 / Cross valid method: THROUGHOUT / ESU R Free: 0.503 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS. THIS ENZYME-INHIBITOR COMPLEX WAS RECENTLY REDETERMINED AT HIGH RESOLUTION IN A DIFFERENT SPACE GROUP (1W7X.PDB). THIS ORIGINAL STRUCTURE ...Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS. THIS ENZYME-INHIBITOR COMPLEX WAS RECENTLY REDETERMINED AT HIGH RESOLUTION IN A DIFFERENT SPACE GROUP (1W7X.PDB). THIS ORIGINAL STRUCTURE IS AT LOW RESOLUTION AND THE DATA IS INCOMPLETE. NEVERTHELESS THE INHIBITOR BIND MODE IS WELL DETERMINED. CURIOUSLY ONE INHIBITOR SUBSTITUENT IN THIS STRUCTURE IS DISPLACED FROM ITS POSITION SEEN IN RELATED STRUCTURES, AND RESIDUES FROM THE NEXT MOLECULE IN THE CRYSTAL FILL THE BINDING POCKET. THE LOOP 140-150 IS NOT SEEN, AND POSSIBLY SUFFERED PROTEOLYSIS.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 19.41 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 3→72.36 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|