 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1dva: Crystal Structure of the Complex Between the Peptide Exosite Inhi... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1dva | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Crystal Structure of the Complex Between the Peptide Exosite Inhibitor E-76 and Coagulation Factor VIIA | |||||||||
 Components Components |
| |||||||||
 Keywords Keywords | HYDROLASE/HYDROLASE INHIBITOR / protein-peptide complex / HYDROLASE-HYDROLASE INHIBITOR COMPLEX | |||||||||
| Function / homology |  Function and homology information Function and homology informationcoagulation factor VIIa / response to Thyroid stimulating hormone / response to astaxanthin / response to thyrotropin-releasing hormone / response to 2,3,7,8-tetrachlorodibenzodioxine / response to carbon dioxide / serine-type peptidase complex / response to genistein / response to vitamin K / positive regulation of platelet-derived growth factor receptor signaling pathway ...coagulation factor VIIa / response to Thyroid stimulating hormone / response to astaxanthin / response to thyrotropin-releasing hormone / response to 2,3,7,8-tetrachlorodibenzodioxine / response to carbon dioxide / serine-type peptidase complex / response to genistein / response to vitamin K / positive regulation of platelet-derived growth factor receptor signaling pathway / positive regulation of leukocyte chemotaxis / response to thyroxine / response to cholesterol / positive regulation of positive chemotaxis / : / response to growth hormone / positive regulation of blood coagulation / animal organ regeneration / positive regulation of TOR signaling / Transport of gamma-carboxylated protein precursors from the endoplasmic reticulum to the Golgi apparatus / Gamma-carboxylation of protein precursors / Removal of aminoterminal propeptides from gamma-carboxylated proteins / BMAL1:CLOCK,NPAS2 activates circadian expression / serine-type peptidase activity / circadian rhythm / protein processing / response to estrogen / Golgi lumen / blood coagulation / response to estradiol / extracellular matrix / vesicle / response to hypoxia / positive regulation of cell migration / endoplasmic reticulum lumen / signaling receptor binding / serine-type endopeptidase activity / calcium ion binding / : / extracellular region / plasma membrane Similarity search - Function | |||||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | |||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / Resolution: 3 Å X-RAY DIFFRACTION / SYNCHROTRON / Resolution: 3 Å | |||||||||
 Authors Authors | Eigenbrot, C. / Ultsch, M.H. | |||||||||
 Citation Citation | Journal: Nature / Year: 2000 Title: Peptide exosite inhibitors of factor VIIa as anticoagulants. Authors: Dennis, M.S. / Eigenbrot, C. / Skelton, N.J. / Ultsch, M.H. / Santell, L. / Dwyer, M.A. / O'Connell, M.P. / Lazarus, R.A. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1dva.cif.gz | 168 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1dva.ent.gz | 128.4 KB | Display | PDB format |
| PDBx/mmJSON format | 1dva.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/dv/1dvaftp://data.pdbj.org/pub/pdb/validation_reports/dv/1dva | HTTPS FTP |
|---|
-Related structure data
| Related structure data | |
|---|---|
| Similar structure data |












-Links
 PDBj
PDBj













- Assembly
Assembly
| Deposited unit | 
| |||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| |||||||||||||||||||||||||||||||||
| 2 | 
| |||||||||||||||||||||||||||||||||
| Unit cell |
| |||||||||||||||||||||||||||||||||
| Noncrystallographic symmetry (NCS) | NCS domain:
|
-Components
-DES-GLA FACTOR VIIA ... , 2 types, 4 molecules HILM
| #1: Protein | Mass: 28103.256 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Organ: LIVER / Plasmid: PCMV5 / Cell line (production host): HUMAN KIDNEY CELL LINE 293 / Production host: Homo sapiens (human) / References: UniProt: P08709, coagulation factor VIIa#2: Protein | Mass: 10994.179 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Organ: LIVER / Plasmid: PCMV5 / Cell line (production host): HUMAN KIDNEY CELL LINE 293 / Production host: Homo sapiens (human) / References: UniProt: P08709, coagulation factor VIIa |
|---|
-Protein/peptide , 1 types, 2 molecules XY
| #3: Protein/peptide | Mass: 2198.461 Da / Num. of mol.: 2 / Source method: obtained synthetically Details: Peptide E-76 was synthesized on a solid support, then cleaved and purified |
|---|
-Sugars , 4 types, 5 molecules 
| #4: Polysaccharide | beta-D-galactopyranose-(1-4)-beta-D-glucopyranose / beta-lactose  Source method: isolated from a genetically manipulated source Details: oligosaccharide / References: beta-lactose | ||||
|---|---|---|---|---|---|
| #8: Sugar |  Type: L-saccharide, alpha linking / Mass: 164.156 Da / Num. of mol.: 2 Type: L-saccharide, alpha linking / Mass: 164.156 Da / Num. of mol.: 2Source method: isolated from a genetically manipulated source Formula: C6H12O5 #9: Sugar | ChemComp-GLC / |  Type: D-saccharide, alpha linking / Mass: 180.156 Da / Num. of mol.: 1 Type: D-saccharide, alpha linking / Mass: 180.156 Da / Num. of mol.: 1Source method: isolated from a genetically manipulated source Formula: C6H12O6 #10: Sugar | ChemComp-FUL / |  Type: L-saccharide, beta linking / Mass: 164.156 Da / Num. of mol.: 1 Type: L-saccharide, beta linking / Mass: 164.156 Da / Num. of mol.: 1Source method: isolated from a genetically manipulated source Formula: C6H12O5 |
-Non-polymers , 4 types, 12 molecules 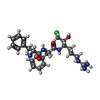

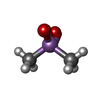

| #5: Chemical |  Type: peptide-like, Peptide-like / Class: Inhibitor / Mass: 504.045 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C25H36ClN6O3 / References: D-Phe-Phe-Arg Chloromethylketone Type: peptide-like, Peptide-like / Class: Inhibitor / Mass: 504.045 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C25H36ClN6O3 / References: D-Phe-Phe-Arg Chloromethylketone#6: Chemical | ChemComp-CA /  Mass: 40.078 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Ca Mass: 40.078 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Ca#7: Chemical |  Mass: 136.989 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C2H6AsO2 Mass: 136.989 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C2H6AsO2#11: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 4 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Details
| Has protein modification | Y |
|---|---|
| Nonpolymer details | THE INHIBITOR IS BOUND TO THE ACTIVE SITE OF THE ENZYME. THE UNBOUND FORM OF THE INHIBITOR IS D-PHE- ...THE INHIBITOR IS BOUND TO THE ACTIVE SITE OF THE ENZYME. THE UNBOUND FORM OF THE INHIBITOR IS D-PHE-PHE-ARG-CHLOROMETH |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.57 Å3/Da / Density % sol: 52.18 % | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 292 K / Method: vapor diffusion, sitting drop / pH: 5.5 Details: PEG4000, t-butanol, sodium cacodylate, pH 5.5, VAPOR DIFFUSION, SITTING DROP, temperature 292K | ||||||||||||||||||||
| Crystal grow | *PLUS Method: vapor diffusion, hanging drop | ||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 110 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SSRL  / Beamline: BL9-1 / Wavelength: 0.98 / Beamline: BL9-1 / Wavelength: 0.98 |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE / Date: Jul 9, 1998 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.98 Å / Relative weight: 1 |
| Reflection | Resolution: 3→50 Å / Num. all: 16915 / Num. obs: 16915 / % possible obs: 97.5 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Redundancy: 2.9 % / Biso Wilson estimate: 56 Å2 / Rmerge(I) obs: 0.104 / Net I/σ(I): 6.2 |
| Reflection shell | Resolution: 3→3.16 Å / Redundancy: 2.9 % / Rmerge(I) obs: 0.262 / Mean I/σ(I) obs: 2.7 / Num. unique all: 2478 / % possible all: 98.4 |
| Reflection | *PLUS |
| Reflection shell | *PLUS % possible obs: 99 % |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Resolution: 3→50 Å / Rfactor Rfree error: 0.011 / Data cutoff high absF: 100000 / Data cutoff low absF: 0.1 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0.2 / σ(I): 0 / Stereochemistry target values: Engh & Huber Details: BULK SOLVENT MODEL WAS APPLIED THERE IS UNPUBLISHED EXPERIMENTAL EVIDENCE THAT THE CARBOHYDRATE ATTACHED TO CHAINS L AND M DIFFERS FROM THAT DESCRIBED IN THIS ENTRY. SER 52 CARRIES 2 OR 3 ...Details: BULK SOLVENT MODEL WAS APPLIED THERE IS UNPUBLISHED EXPERIMENTAL EVIDENCE THAT THE CARBOHYDRATE ATTACHED TO CHAINS L AND M DIFFERS FROM THAT DESCRIBED IN THIS ENTRY. SER 52 CARRIES 2 OR 3 GLUCOSE RESIDUES, AND SER 60 CARRIES ALPHA-L-FUCOSE. THE ELECTRON DENSITY IN THIS REGION IS IMPERFECT, AND WAS FIT WITHOUT THIS INFORMATION. THE FIT IS ONLY MODERATELY SUCCESFUL.
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 49.2 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 3→50 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints NCS | Refine-ID: X-RAY DIFFRACTION / Weight Biso : 1 / Weight position: 200
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 3→3.19 Å / Rfactor Rfree error: 0.037 / Total num. of bins used: 6
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: X-PLOR / Version: 3.851 / Classification: refinement | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS σ(F): 0.2 / % reflection Rfree: 3.9 % / Rfactor obs: 0.225 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS Biso mean: 49.2 Å2 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Rfactor Rfree: 0.361 / % reflection Rfree: 3.3 % / Rfactor Rwork: 0.35 |
