 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1qo0: Amide receptor of the amidase operon of Pseudomonas aeruginosa (A... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1qo0 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Amide receptor of the amidase operon of Pseudomonas aeruginosa (AmiC) complexed with the negative regulator AmiR. | ||||||
 Components Components |
| ||||||
 Keywords Keywords | BINDING PROTEIN / GENE REGULATOR / RECEPTOR | ||||||
| Function / homology |  Function and homology information Function and homology information: / : / carbon utilization / amino acid transport / transcription antitermination / kinase activity / RNA binding Similarity search - Function | ||||||
| Biological species |   PSEUDOMONAS AERUGINOSA (bacteria) PSEUDOMONAS AERUGINOSA (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.25 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.25 Å | ||||||
 Authors Authors | Pearl, L.H. / O'Hara, B.P. / Roe, S.M. | ||||||
 Citation Citation | Journal: Embo J. / Year: 1999 Title: Crystal Structure and Induction Mechanism of Amic-Amir: A Ligand-Regulated Transcription Antitermination Complex Authors: O'Hara, B.P. / Norman, R.A. / Wan, P.T.C. / Roe, S.M. / Barrett, T.E. / Drew, R.E. / Pearl, L.H. #1: Journal: Embo J. / Year: 1994Title: Crystal Structure of Amic: The Controller of Transcription Antitermination in the Amidase Operon of Pseudomonas Aeruginosa Authors: Pearl, L.H. / O'Hara, B.P. / Drew, R.E. / Wilson, S.W. #2: Journal: Embo J. / Year: 1993 Title: Antitermination of Amidase Expression in Pseudomonas Aeruginosa is Controlled by a Novel Cytoplasmic Amide- Binding Protein Authors: Wilson, S.A. / Wachira, S.J. / Drew, R.E. / Jones, D. / Pearl, L.H. #3: Journal: J.Mol.Biol. / Year: 1991 Title: Crystallization and Preliminary X-Ray Data for the Negative Regulator (Amic) of the Amidase Operon of Pseudomonas Aeruginosa Authors: Wilson, S.A. / Chayen, N.E. / Hemmings, A.M. / Drew, R.E. / Pearl, L.H. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1qo0.cif.gz | 246.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1qo0.ent.gz | 196.7 KB | Display | PDB format |
| PDBx/mmJSON format | 1qo0.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/qo/1qo0ftp://data.pdbj.org/pub/pdb/validation_reports/qo/1qo0 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1peaS S: Starting model for refinement |
|---|---|
| Similar structure data |







-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 42960.031 Da / Num. of mol.: 2 / Fragment: AMIDE RECEPTOR Source method: isolated from a genetically manipulated source Details: COMPLEXED WITH BUTYRAMIDE MOLECULE / Source: (gene. exp.) PSEUDOMONAS AERUGINOSA (bacteria) / Strain: PAC1 / Production host: PSEUDOMONAS AERUGINOSA (bacteria) / References: UniProt: P27017#2: Protein | Mass: 21903.287 Da / Num. of mol.: 2 / Fragment: AMIDE RECEPTOR/NEGATIVE REGULATOR Source method: isolated from a genetically manipulated source Source: (gene. exp.) PSEUDOMONAS AERUGINOSA (bacteria) / Strain: PAC1 / Production host: PSEUDOMONAS AERUGINOSA (bacteria) / References: UniProt: P10932#3: Chemical | 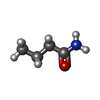  Mass: 87.120 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C4H9NO Mass: 87.120 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C4H9NO#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 851 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 851 / Source method: isolated from a natural source / Formula: H2OSequence details | SWISSPROT HAS THE WRONG SEQUENCE, STRUCTURE IS CORRECT. CHAINS A AND B GLN 27 SWS P27017 HIS 26 ARG ...SWISSPROT HAS THE WRONG SEQUENCE, STRUCTURE IS CORRECT. CHAINS A AND B GLN 27 SWS P27017 HIS 26 ARG 28 SWS P27017 ALA 27 CHAINS D AND E ARG 64 SWS P10932 GLY 27 | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.96 Å3/Da / Density % sol: 58 % |
|---|---|
| Crystal grow | Method: microbatch / pH: 5.6 Details: CRYSTALLISATION WAS BY MICRO-BATCH AND STREAK SEEDING. WELLS CONTAINED 8-8.5% PEG4000, 20%(V/V) 2-PROPANOL, 50MM SODIUM CITRATE BUFFERED AT PH 5.6 AND AMIC-AMIR COMPLEX AT 5MG/ML (FINAL CONCENTRATION). |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SRS  / Beamline: PX7.2 / Wavelength: 1.488 / Beamline: PX7.2 / Wavelength: 1.488 |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE / Date: Jul 15, 1998 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.488 Å / Relative weight: 1 |
| Reflection | Resolution: 2.25→33.6 Å / Num. obs: 67099 / % possible obs: 83.8 % / Redundancy: 2.7 % / Biso Wilson estimate: 23.6 Å2 / Rsym value: 0.052 / Net I/σ(I): 6.8 |
| Reflection shell | Resolution: 2.25→2.36 Å / Redundancy: 1.5 % / Mean I/σ(I) obs: 3 / Rsym value: 0.177 / % possible all: 69.3 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1PEA Resolution: 2.25→20 Å / SU B: 6.63 / SU ML: 0.16 / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R: 0.31 / ESU R Free: 0.24
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 9.48 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.25→20 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|
