 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1qnl: AMIDE RECEPTOR/NEGATIVE REGULATOR OF THE AMIDASE OPERON OF PSEUDO... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1qnl | ||||||
|---|---|---|---|---|---|---|---|
| Title | AMIDE RECEPTOR/NEGATIVE REGULATOR OF THE AMIDASE OPERON OF PSEUDOMONAS AERUGINOSA (AMIC) COMPLEXED WITH BUTYRAMIDE | ||||||
 Components Components | ALIPHATIC AMIDASE EXPRESSION-REGULATING PROTEIN | ||||||
 Keywords Keywords | TRANSFERASE / BINDING PROTEIN / GENE REGULATOR / RECEPTOR / KINASE / REPRESSOR | ||||||
| Function / homology |  Function and homology information Function and homology information: / : / negative regulation of hydrolase activity / amino acid transport / kinase activity Similarity search - Function | ||||||
| Biological species |   PSEUDOMONAS AERUGINOSA (bacteria) PSEUDOMONAS AERUGINOSA (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.7 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.7 Å | ||||||
 Authors Authors | Pearl, L.H. / O'Hara, B.P. / Roe, S.M. | ||||||
 Citation Citation | Journal: Protein Eng. / Year: 2000 Title: Structural Adaptation to Selective Pressure for Altered Ligand Specificity in the Pseudomonas Aeruginosa Amide Receptor, Amic Authors: O'Hara, B.P. / Wilson, S.A. / Lee, A.W.L. / Roe, S.M. / Siligardi, G. / Drew, R.E. / Pearl, L.H. #1: Journal: Embo J. / Year: 1994Title: Crystal Structure of Amic: The Controller of Transcription Antitermination in the Amidase Operon of Pseudomonas Aeruginosa Authors: Pearl, L.H. / O'Hara, B.P. / Drew, R.E. / Wilson, S.A. #2: Journal: Embo J. / Year: 1993 Title: Antitermination of Amidase Expression in Pseudomonas Aeruginosa is Controlled by a Novel Cytoplasmic Amide- Binding Protein Authors: Wilson, S.A. / Wachira, S.J. / Drew, R.E. / Jones, D. / Pearl, L.H. #3: Journal: J.Mol.Biol. / Year: 1991 Title: Crystallization and Preliminary X-Ray Data for the Negative Regulator (Amic) of the Amidase Operon of Pseudomonas Aeruginosa Authors: Wilson, S.A. / Chayen, N.E. / Hemmings, A.M. / Drew, R.E. / Pearl, L.H. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1qnl.cif.gz | 90 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1qnl.ent.gz | 67.2 KB | Display | PDB format |
| PDBx/mmJSON format | 1qnl.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/qn/1qnlftp://data.pdbj.org/pub/pdb/validation_reports/qn/1qnl | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1peaS S: Starting model for refinement |
|---|---|
| Similar structure data |








-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 42973.027 Da / Num. of mol.: 1 / Fragment: AMIDE RECEPTOR/NEGATIVE REGULATOR, RESIDUES 7-374 Source method: isolated from a genetically manipulated source Details: BUTYRAMIDE COMPLEX / Source: (gene. exp.) PSEUDOMONAS AERUGINOSA (bacteria) / Strain: PAC1 / Production host: PSEUDOMONAS AERUGINOSA (bacteria) / References: UniProt: P27017 |
|---|---|
| #2: Chemical | ChemComp-BMD / 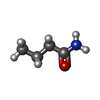  Mass: 87.120 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H9NO Mass: 87.120 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H9NO |
| #3: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 43 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 43 / Source method: isolated from a natural source / Formula: H2O |
| Sequence details | SWISSPROT HAS THE WRONG SEQUENCE, STRUCTURE IS CORRECT. GLN 27 UNP P27017 HIS 26 ARG 28 UNP P27017 ALA 27 |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.16 Å3/Da / Density % sol: 43 % |
|---|---|
| Crystal grow | Method: microbatch / pH: 7 Details: MICROBATCH CRYSTALLISATION. WELLS CONTAIN 12.8MG/ML PAC181-AMIC, 5MM BUTYRAMIDE, 1.36M NA CITRATE, 100MM HEPES-NAOH PH7.5, pH 7.00 |
| Crystal grow | *PLUS Method: other / Details: Chayen, N.E., (1992) J. Crystal Growth, 122, 176. |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RUH2R / Wavelength: 1.5418 |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE / Date: Dec 15, 1996 / Details: MIRRORS |
| Radiation | Monochromator: NI FILTER / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 2.7→38 Å / Num. obs: 10389 / % possible obs: 97.8 % / Observed criterion σ(I): 1 / Redundancy: 2.7 % / Biso Wilson estimate: 54.2 Å2 / Rmerge(I) obs: 0.098 / Net I/σ(I): 4.5 |
| Reflection shell | Resolution: 2.7→2.84 Å / Redundancy: 2.6 % / Rmerge(I) obs: 0.185 / Mean I/σ(I) obs: 3.8 / % possible all: 97.2 |
| Reflection shell | *PLUS % possible obs: 97.2 % |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1PEA Resolution: 2.7→15 Å / SU B: 16.04 / SU ML: 0.34 / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R Free: 0.47 Details: RESIDUES 2 - 7, 40 - 50, 222 - 226, 347 - 349 AND 376 - 385 IN THIS MOLECULE ARE NOT DEFINED IN THE ELECTRON DENSITY AND ARE PRESUMED TO BE DISORDERED. THERE ARE ALSO A FEW OTHER RESIDUES ...Details: RESIDUES 2 - 7, 40 - 50, 222 - 226, 347 - 349 AND 376 - 385 IN THIS MOLECULE ARE NOT DEFINED IN THE ELECTRON DENSITY AND ARE PRESUMED TO BE DISORDERED. THERE ARE ALSO A FEW OTHER RESIDUES THAT COULD NOT BE SATISFACTORILY REFINED, NAMELY: 37, 68, 72, 74, 137, 165, 194, 195, 261, 307, 318, 319, 328, 359. SOME OF THIS ARE MISSING MAIN CHAIN AS WELL AS SIDE CHAIN ATOMS.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 27.38 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.7→15 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: REFMAC / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Rfactor obs: 0.24 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS |
