 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
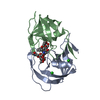
Yorodumi- PDB-1c6z: ALTERNATE BINDING SITE FOR THE P1-P3 GROUP OF A CLASS OF POTENT H... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1c6z | ||||||
|---|---|---|---|---|---|---|---|
| Title | ALTERNATE BINDING SITE FOR THE P1-P3 GROUP OF A CLASS OF POTENT HIV-1 PROTEASE INHIBITORS AS A RESULT OF CONCERTED STRUCTURAL CHANGE IN 80'S LOOP. | ||||||
 Components Components | PROTEIN (PROTEASE) | ||||||
 Keywords Keywords | HYDROLASE/HYDROLASE INHIBITOR / hydrolase-hydrolase inhibitor complex | ||||||
| Function / homology |  Function and homology information Function and homology informationhost multivesicular body / aspartic-type endopeptidase activity / virion membrane / proteolysis Similarity search - Function | ||||||
| Biological species |   Human immunodeficiency virus 1 Human immunodeficiency virus 1 | ||||||
| Method |  X-RAY DIFFRACTION / Resolution: 2.5 Å X-RAY DIFFRACTION / Resolution: 2.5 Å | ||||||
 Authors Authors | Munshi, S. | ||||||
 Citation Citation | Journal: Acta Crystallogr.,Sect.D / Year: 2000 Title: An alternate binding site for the P1-P3 group of a class of potent HIV-1 protease inhibitors as a result of concerted structural change in the 80s loop of the protease. Authors: Munshi, S. / Chen, Z. / Yan, Y. / Li, Y. / Olsen, D.B. / Schock, H.B. / Galvin, B.B. / Dorsey, B. / Kuo, L.C. #1: Journal: J.Med.Chem. / Year: 1991Title: Novel Binding Mode of Highly Potent HIV-Proteinase Inhibitors Incorporating the (R)-Hydroxyethylamine Isostere Authors: Krohn, A. / Redshaw, S. / Ritchie, J.C. / Graves, B.J. / Hatada, M.H. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1c6z.cif.gz | 53.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1c6z.ent.gz | 39.8 KB | Display | PDB format |
| PDBx/mmJSON format | 1c6z.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/c6/1c6zftp://data.pdbj.org/pub/pdb/validation_reports/c6/1c6z | HTTPS FTP |
|---|
-Related structure data






















-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 10801.674 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Details: NY5 ISOLATE / Source: (gene. exp.) Human immunodeficiency virus 1 / Genus: Lentivirus / Gene: NY5 ISOLATE / Production host:  References: UniProt: O09893, Hydrolases; Acting on peptide bonds (peptidases); Metalloendopeptidases #2: Chemical | ChemComp-ROC / ( | 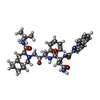  Type: peptide-like, Peptide-like / Class: Inhibitor / Mass: 670.841 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C38H50N6O5 / References: Saquinavir / Comment: medication, antiretroviral*YM Type: peptide-like, Peptide-like / Class: Inhibitor / Mass: 670.841 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C38H50N6O5 / References: Saquinavir / Comment: medication, antiretroviral*YM#3: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 76 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 76 / Source method: isolated from a natural source / Formula: H2OCompound details | 9X MUTANT PROTEASE HAS NINE POINT MUTATIONS COMPARED TO WILD TYPE: ...9X MUTANT PROTEASE HAS NINE POINT MUTATIONS COMPARED TO WILD TYPE: L10V,K20M,L24I,S37D,M46I,I54V,L63P,A71V,V82T THERE IS ONE PROTEASE DIMER IN AN ASYMMETRIC | Nonpolymer details | THE INHIBITOR ROC IS A HYDROXYETH | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.96 Å3/Da / Density % sol: 58.5 % | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Details: CRYSTALS OBTAINED BY CO-CRYSTALLIZATION AT PH 5.2, USING 0.6M NaCl AS PRECIPITATING AGENT IN 0.1M SODIUM ACETATE BUFFER. PROTEIN WAS AT 5.5 MG/ML CONCENTRATION. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS pH: 5 / Method: vapor diffusion, hanging dropDetails: inhibitor-protein mixture and the reservoir solution were mixed in a 1:1(v/v) ratio | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
|---|---|
| Radiation wavelength | Relative weight: 1 |
| Reflection | Num. all: 41654 / Num. obs: 7717 / % possible obs: 82 % / Rmerge(I) obs: 0.062 |
| Reflection shell | Resolution: 2.5→2.59 Å / % possible all: 73 |
| Reflection | *PLUS Highest resolution: 2.5 Å / Lowest resolution: 20 Å / Num. measured all: 41654 |
| Reflection shell | *PLUS % possible obs: 73 % / Num. unique obs: 677 / Num. measured obs: 2345 / Rmerge(I) obs: 0.224 / Mean I/σ(I) obs: 4 |
- Processing
Processing
| Software | Name: X-PLOR / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Resolution: 2.5→8 Å / Rfactor Rfree: 0.29 / Rfactor Rwork: 0.18 / Rfactor obs: 0.18 / σ(F): 2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.5→8 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|
