 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-5ahc: Disubstituted bis-THF moieties as new P2 ligands in non-peptidal ... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 5ahc | ||||||
|---|---|---|---|---|---|---|---|
| Title | Disubstituted bis-THF moieties as new P2 ligands in non-peptidal HIV- 1 Protease Inhibitors (II) | ||||||
 Components Components | PROTEASE | ||||||
 Keywords Keywords | HYDROLASE / INHIBITOR / RATIONAL DRUG DESIGN BIS-THF BIS-DIOL | ||||||
| Function / homology |  Function and homology information Function and homology informationHIV-1 retropepsin / symbiont-mediated activation of host apoptosis / retroviral ribonuclease H / exoribonuclease H / exoribonuclease H activity / DNA integration / viral genome integration into host DNA / establishment of integrated proviral latency / RNA-directed DNA polymerase / RNA stem-loop binding ...HIV-1 retropepsin / symbiont-mediated activation of host apoptosis / retroviral ribonuclease H / exoribonuclease H / exoribonuclease H activity / DNA integration / viral genome integration into host DNA / establishment of integrated proviral latency / RNA-directed DNA polymerase / RNA stem-loop binding / viral penetration into host nucleus / host multivesicular body / RNA-directed DNA polymerase activity / RNA-DNA hybrid ribonuclease activity / Transferases; Transferring phosphorus-containing groups; Nucleotidyltransferases / host cell / viral nucleocapsid / DNA recombination / DNA-directed DNA polymerase / aspartic-type endopeptidase activity / Hydrolases; Acting on ester bonds / DNA-directed DNA polymerase activity / symbiont-mediated suppression of host gene expression / viral translational frameshifting / symbiont entry into host cell / lipid binding / host cell nucleus / host cell plasma membrane / virion membrane / structural molecule activity / proteolysis / DNA binding / zinc ion binding Similarity search - Function | ||||||
| Biological species |   HUMAN IMMUNODEFICIENCY VIRUS TYPE 1 HUMAN IMMUNODEFICIENCY VIRUS TYPE 1 | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.5 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.5 Å | ||||||
 Authors Authors | Hohlfeld, K. / Wegner, J.K. / Kesteleyn, B. / Linclau, B. / Unge, J. | ||||||
 Citation Citation | Journal: J.Med.Chem. / Year: 2015 Title: Disubstituted Bis-Thf Moieties as New P2 Ligands in Non-Peptidal HIV-1 Protease Inhibitors (II). Authors: Hohlfeld, K. / Wegner, J. / Kesteleyn, B. / Linclau, B. / Unge, J. | ||||||
| History |
| ||||||
| Remark 700 | SHEET DETERMINATION METHOD: DSSP THE SHEETS PRESENTED AS "AB" IN EACH CHAIN ON SHEET RECORDS BELOW ... SHEET DETERMINATION METHOD: DSSP THE SHEETS PRESENTED AS "AB" IN EACH CHAIN ON SHEET RECORDS BELOW IS ACTUALLY AN 6-STRANDED BARREL THIS IS REPRESENTED BY A 7-STRANDED SHEET IN WHICH THE FIRST AND LAST STRANDS ARE IDENTICAL. THE SHEETS PRESENTED AS "BA" IN EACH CHAIN ON SHEET RECORDS BELOW IS ACTUALLY AN 6-STRANDED BARREL THIS IS REPRESENTED BY A 7-STRANDED SHEET IN WHICH THE FIRST AND LAST STRANDS ARE IDENTICAL. |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 5ahc.cif.gz | 60.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb5ahc.ent.gz | 44.2 KB | Display | PDB format |
| PDBx/mmJSON format | 5ahc.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ah/5ahcftp://data.pdbj.org/pub/pdb/validation_reports/ah/5ahc | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  5agzC  5ah6C  5ah7C  5ah8C  5ah9C  5ahaC  5ahbC C: citing same article ( |
|---|---|
| Similar structure data |



















-Links
 PDBj
PDBj


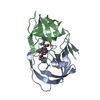
- Assembly
Assembly
| Deposited unit | 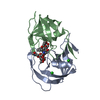
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 10775.659 Da / Num. of mol.: 2 / Fragment: ASPARTYL PROTEASE, RESIDUES 501-599 / Mutation: YES Source method: isolated from a genetically manipulated source Source: (gene. exp.) HUMAN IMMUNODEFICIENCY VIRUS TYPE 1 (Z2/CDC-Z34 ISOLATE)Strain: 99HHP1 (D10) / Plasmid: PEXP5 / Production host:  #2: Chemical |   Mass: 35.453 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: Cl Mass: 35.453 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: Cl#3: Chemical | ChemComp-VXL / ( |   Mass: 649.752 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C31H43N3O10S Mass: 649.752 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C31H43N3O10S#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 277 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 277 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.7 Å3/Da / Density % sol: 54.42 % / Description: NONE |
|---|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: MAX II  / Beamline: I911-3 / Wavelength: 1 / Beamline: I911-3 / Wavelength: 1 |
| Detector | Type: MARMOSAIC 225 mm CCD / Detector: CCD / Date: Apr 17, 2012 / Details: MIRRORS |
| Radiation | Monochromator: SI / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 |
| Reflection | Resolution: 1.5→25.76 Å / Num. obs: 37974 / % possible obs: 99.7 % / Observed criterion σ(I): 4 / Redundancy: 4.8 % / Rmerge(I) obs: 0.11 / Net I/σ(I): 2.6 |
| Reflection shell | Resolution: 1.5→1.58 Å / Redundancy: 4.8 % / Rmerge(I) obs: 0.43 / Mean I/σ(I) obs: 0.5 / % possible all: 100 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: NON-PUBLISHED Resolution: 1.5→25.76 Å / Cor.coef. Fo:Fc: 0.939 / Cor.coef. Fo:Fc free: 0.928 / SU B: 1.257 / SU ML: 0.049 / Cross valid method: THROUGHOUT / ESU R: 0.081 / ESU R Free: 0.08 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS. U VALUES REFINED INDIVIDUALLY
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 15.869 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.5→25.76 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|