 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-4anj: MYOSIN VI (MDinsert2-GFP fusion) PRE-POWERSTROKE STATE (MG.ADP.AlF4) -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4anj | ||||||
|---|---|---|---|---|---|---|---|
| Title | MYOSIN VI (MDinsert2-GFP fusion) PRE-POWERSTROKE STATE (MG.ADP.AlF4) | ||||||
 Components Components |
| ||||||
 Keywords Keywords |  MOTOR PROTEIN/METAL-BINDNG PROTEIN / MOTOR PROTEIN-METAL-BINDNG PROTEIN COMPLEX / MOLECULAR MOTOR / METAL-BINDING PROTEIN / TRANSITION STATE / PRE-POWERSTROKE STATE / GFP FUSION MOTOR PROTEIN/METAL-BINDNG PROTEIN / MOTOR PROTEIN-METAL-BINDNG PROTEIN COMPLEX / MOLECULAR MOTOR / METAL-BINDING PROTEIN / TRANSITION STATE / PRE-POWERSTROKE STATE / GFP FUSION | ||||||
| Function / homology |  Function and homology information Function and homology informationnegative regulation of phospholipase C-activating phototransduction signaling pathway / myosin VI complex / myosin VI head/neck binding / myosin VII complex / photoreceptor cell axon guidance / negative regulation of opsin-mediated signaling pathway / rhabdomere / rhabdomere development / myosin V complex / : ...negative regulation of phospholipase C-activating phototransduction signaling pathway / myosin VI complex / myosin VI head/neck binding / myosin VII complex / photoreceptor cell axon guidance / negative regulation of opsin-mediated signaling pathway / rhabdomere / rhabdomere development / myosin V complex / : / regulation of secretion / kinetochore organization / : / actin filament-based movement / G protein-coupled opsin signaling pathway / Neutrophil degranulation / inner ear auditory receptor cell differentiation / myosin V binding / channel regulator activity / vesicle transport along actin filament / cellular response to ethanol / myosin complex / clathrin-coated vesicle / microfilament motor activity / inner ear morphogenesis / muscle cell cellular homeostasis / myosin heavy chain binding / mitotic spindle pole / filamentous actin / microvillus / centriole replication / cytoskeletal motor activity / DNA damage response, signal transduction by p53 class mediator / enzyme regulator activity / ruffle / centriole / bioluminescence / filopodium / generation of precursor metabolites and energy / actin filament organization / actin filament / ADP binding / sensory perception of sound / intracellular protein transport / mitotic spindle / spindle / ruffle membrane / endocytosis / actin filament binding / sensory perception of smell / actin cytoskeleton / cell cortex / midbody / cytoplasmic vesicle / nuclear membrane / vesicle / calmodulin binding / protein phosphorylation / centrosome / calcium ion binding / perinuclear region of cytoplasm / Golgi apparatus / nucleoplasm / ATP binding / nucleus / cytosol / cytoplasmSimilarity search - Function | ||||||
| Biological species |  SUS SCROFA (pig) SUS SCROFA (pig)  AEQUOREA VICTORIA (jellyfish) AEQUOREA VICTORIA (jellyfish) DROSOPHILA MELANOGASTER (fruit fly) DROSOPHILA MELANOGASTER (fruit fly) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.6 Å | ||||||
 Authors Authors | Menetrey, J. / Isabet, T. / Ropars, V. / Mukherjea, M. / Pylypenko, O. / Liu, X. / Perez, J. / Vachette, P. / Sweeney, H.L. / Houdusse, A.M. | ||||||
 Citation Citation | Journal: Mol.Cell / Year: 2012 Title: Processive Steps in the Reverse Direction Require Uncoupling of the Lead Head Lever Arm of Myosin Vi. Authors: Menetrey, J. / Isabet, T. / Ropars, V. / Mukherjea, M. / Pylypenko, O. / Liu, X. / Perez, J. / Vachette, P. / Sweeney, H.L. / Houdusse, A.M. | ||||||
| History |
| ||||||
| Remark 700 | SHEET DETERMINATION METHOD: DSSP THE SHEETS PRESENTED AS "AH" IN EACH CHAIN ON SHEET RECORDS BELOW ... SHEET DETERMINATION METHOD: DSSP THE SHEETS PRESENTED AS "AH" IN EACH CHAIN ON SHEET RECORDS BELOW IS ACTUALLY AN 11-STRANDED BARREL THIS IS REPRESENTED BY A 12-STRANDED SHEET IN WHICH THE FIRST AND LAST STRANDS ARE IDENTICAL. |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4anj.cif.gz | 238.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4anj.ent.gz | 184 KB | Display | PDB format |
| PDBx/mmJSON format | 4anj.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/an/4anjftp://data.pdbj.org/pub/pdb/validation_reports/an/4anj | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  4e7sC  4e7zC  2v26S  3dqaS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |
























































































-Links
 PDBj
PDBj










- Assembly
Assembly
| Deposited unit | 
| |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| |||||||||
| Unit cell |
| |||||||||
| Components on special symmetry positions |
|
-Components
-Protein , 2 types, 2 molecules AB
| #1: Protein | Mass: 120131.617 Da / Num. of mol.: 1 / Fragment: MYOSIN-6 RESIDUES 1-817, GFP RESIDUES 2-238 Source method: isolated from a genetically manipulated source Details: CHIMERIC PROTEIN, MYOSIN-6 RESIDUES 1-816 (AUTHOR NUMBERING) AND COMPLETE GFP (AFTER INITIATION METHIONINE REMOVAL) Source: (gene. exp.) SUS SCROFA (pig), (gene. exp.) AEQUOREA VICTORIA (jellyfish)Cell line (production host): SF9 / Production host:  SPODOPTERA FRUGIPERDA (fall armyworm) / References: UniProt: Q29122, UniProt: P42212 SPODOPTERA FRUGIPERDA (fall armyworm) / References: UniProt: Q29122, UniProt: P42212 |
|---|---|
| #2: Protein | / CAM Mass: 16825.520 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) DROSOPHILA MELANOGASTER (fruit fly) / Cell line (production host): SF9 / Production host: SPODOPTERA FRUGIPERDA (fall armyworm) / References: UniProt: P62152 |
-Non-polymers , 5 types, 97 molecules 

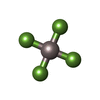


| #3: Chemical | ChemComp-ADP / Adenosine diphosphate Mass: 427.201 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H15N5O10P2 / Comment: ADP, energy-carrying molecule*YM Mass: 427.201 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H15N5O10P2 / Comment: ADP, energy-carrying molecule*YM | ||
|---|---|---|---|
| #4: Chemical | ChemComp-MG /  Mass: 24.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mg Mass: 24.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mg | ||
| #5: Chemical | ChemComp-ALF /  Mass: 102.975 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: AlF4 Mass: 102.975 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: AlF4 | ||
| #6: Chemical |  Mass: 40.078 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: Ca Mass: 40.078 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: Ca#7: Water | ChemComp-HOH / | WaterMass: 18.015 Da / Num. of mol.: 91 / Source method: isolated from a natural source / Formula: H2O |
-Details
| Sequence details | GFP FUSED TO MYOSIN VI, THE AUTHORS STATE THAT THE ORIGINAL SEQUENCE (UNIPROT Q29122) OF MYOSIN VI ...GFP FUSED TO MYOSIN VI, THE AUTHORS STATE THAT THE ORIGINAL SEQUENCE (UNIPROT Q29122) OF MYOSIN VI FROM PIG WAS MOST LIKELY INCORRECT BECAUSE THE CHANGES THAT ARE IN THEIR CLONE (LYS DELETION AND THE 6 MUTATIONS) ARE CONSERVED ACROSS THE MYOSIN VI FAMILY. |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.04 Å3/Da / Density % sol: 59.54 % / Description: NONE |
|---|---|
| Crystal grow | Details: 5% PEG 8000, 50 MM MES PH 5.5, 100 MM NH4SO4, 20 MM MGCL2, 20 MM NACL AND 3 % PROPAN-2-OL |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SOLEIL  / Beamline: PROXIMA 1 / Wavelength: 0.9334 / Beamline: PROXIMA 1 / Wavelength: 0.9334 |
| Detector | Type: ADSC QUANTUM 315r / Detector: CCD / Date: Apr 15, 2009 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9334 Å / Relative weight: 1 |
| Reflection | Resolution: 2.6→48.8 Å / Num. obs: 51178 / % possible obs: 100 % / Observed criterion σ(I): 2 / Redundancy: 4.8 % / Rmerge(I) obs: 0.11 / Net I/σ(I): 7.2 |
| Reflection shell | Resolution: 2.6→48.8 Å / Redundancy: 4.8 % / Rmerge(I) obs: 0.43 / Mean I/σ(I) obs: 2 / % possible all: 100 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRIES 2V26, 3DQA Resolution: 2.6→137.36 Å / Cor.coef. Fo:Fc: 0.889 / Cor.coef. Fo:Fc free: 0.84 / SU B: 11.08 / SU ML: 0.242 / Cross valid method: THROUGHOUT / ESU R: 0.517 / ESU R Free: 0.325 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS SIDE-CHAINS. THAT HAVE NO DEFINED ELECTRON DENSITY WERE OMITTED FROM THE MODEL. REGIONS 35-38, 175-177, 355-361, 395-407 AND 623-638, FROM ...Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS SIDE-CHAINS. THAT HAVE NO DEFINED ELECTRON DENSITY WERE OMITTED FROM THE MODEL. REGIONS 35-38, 175-177, 355-361, 395-407 AND 623-638, FROM THE MYOSIN VI (CHAIN A) AND REGIONS 27-30, 76- 79 AND 111- 116 FROM THE CALMODULIN (CHAIN B) HAVE NO DEFINED ELECTRON DENSITY AND WERE OMITTED FROM THE MODEL.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 23.874 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.6→137.36 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|