 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3r7w | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal Structure of Gtr1p-Gtr2p complex | ||||||
 Components Components |
| ||||||
 Keywords Keywords |  PROTEIN TRANSPORT / Rag GTPases / Gtr1p / Gtr2p / mTOR PROTEIN TRANSPORT / Rag GTPases / Gtr1p / Gtr2p / mTOR | ||||||
| Function / homology |  Function and homology information Function and homology informationMTOR signalling / protein localization to vacuolar membrane / Gtr1-Gtr2 GTPase complex / Ragulator complex / phosphate ion transport / microautophagy / Amino acids regulate mTORC1 / endocytic recycling / fungal-type vacuole membrane / transcription by RNA polymerase I ...MTOR signalling / protein localization to vacuolar membrane / Gtr1-Gtr2 GTPase complex / Ragulator complex / phosphate ion transport / microautophagy / Amino acids regulate mTORC1 / endocytic recycling / fungal-type vacuole membrane / transcription by RNA polymerase I / transcription by RNA polymerase III / positive regulation of TOR signaling / subtelomeric heterochromatin formation / positive regulation of TORC1 signaling / cellular response to starvation / negative regulation of autophagy / Hydrolases; Acting on acid anhydrides; Acting on GTP to facilitate cellular and subcellular movement / late endosome / late endosome membrane / chromosome, telomeric region / GTPase activity / chromatin / GTP binding / positive regulation of transcription by RNA polymerase II / identical protein binding / nucleus / cytoplasmSimilarity search - Function | ||||||
| Biological species |  Saccharomyces cerevisiae (brewer's yeast) Saccharomyces cerevisiae (brewer's yeast) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / SAD / Resolution: 2.773 Å | ||||||
 Authors Authors | Gong, R. / Li, L. / Liu, Y. / Wang, P. / Yang, H. / Wang, L. / Cheng, J. / Guan, K.L. / Xu, Y. | ||||||
 Citation Citation | Journal: Genes Dev. / Year: 2011 Title: Crystal structure of the Gtr1p-Gtr2p complex reveals new insights into the amino acid-induced TORC1 activation Authors: Gong, R. / Li, L. / Liu, Y. / Wang, P. / Yang, H. / Wang, L. / Cheng, J. / Guan, K.L. / Xu, Y. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3r7w.cif.gz | 234.9 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3r7w.ent.gz | 196.7 KB | Display | PDB format |
| PDBx/mmJSON format | 3r7w.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/r7/3r7wftp://data.pdbj.org/pub/pdb/validation_reports/r7/3r7w | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|










-Links
 PDBj
PDBj




- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 36036.566 Da / Num. of mol.: 2 / Fragment: residues 8-310 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Saccharomyces cerevisiae (brewer's yeast)Gene: GTR1 / Plasmid: pETDuet1 / Production host:  Escherichia coli (E. coli) / Strain (production host): BL21(DE3) / References: UniProt: Q00582 Escherichia coli (E. coli) / Strain (production host): BL21(DE3) / References: UniProt: Q00582#2: Protein | Mass: 38067.758 Da / Num. of mol.: 2 / Fragment: residues 11-341 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Saccharomyces cerevisiae (brewer's yeast)Gene: GTR2 / Plasmid: pETDuet1 / Production host: Escherichia coli (E. coli) / Strain (production host): BL21(DE3) / References: UniProt: P53290#3: Chemical | ChemComp-GNP / 5'-Guanylyl imidodiphosphate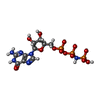  Mass: 522.196 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C10H17N6O13P3 Mass: 522.196 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C10H17N6O13P3Comment: GppNHp, GMPPNP, energy-carrying molecule analogue*YM #4: Chemical | ChemComp-MG /   Mass: 24.305 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Mg Mass: 24.305 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Mg#5: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 4 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 4 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.34 Å3/Da / Density % sol: 63.19 % |
|---|---|
| Crystal grow | Temperature: 277 K / Method: vapor diffusion, hanging drop / pH: 7 Details: 0.1M HEPES, 10% PEG monomethyl ether 5000, 5%(v/v) Tacsimate, pH 7.0, VAPOR DIFFUSION, HANGING DROP, temperature 277K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SSRF  / Beamline: BL17U / Wavelength: 0.97916 Å / Beamline: BL17U / Wavelength: 0.97916 Å |
| Detector | Type: MARMOSAIC 225 mm CCD / Detector: CCD / Date: May 6, 2010 |
| Radiation | Monochromator: SAGITALLY FOCUSED Si(111) / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.97916 Å / Relative weight: 1 |
| Reflection | Resolution: 2.77→50 Å / Num. obs: 46969 / % possible obs: 95.1 % / Biso Wilson estimate: 73.31 Å2 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: SAD / Resolution: 2.773→44.06 Å / Occupancy max: 1 / Occupancy min: 1 / SU ML: 0.37 / σ(F): 1.34 / Phase error: 36.08 / Stereochemistry target values: ML
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.72 Å / VDW probe radii: 1 Å / Solvent model: FLAT BULK SOLVENT MODEL / Bsol: 44.489 Å2 / ksol: 0.284 e/Å3 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 228.52 Å2 / Biso mean: 97.2195 Å2 / Biso min: 40.6 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.773→44.06 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION / Total num. of bins used: 16
|
