 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2wur | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Atomic resolution structure of GFP measured on a rotating anode | |||||||||
 Components Components | GREEN FLUORESCENT PROTEIN | |||||||||
 Keywords Keywords | FLUORESCENT PROTEIN / CHROMOPHORE / BETA-BARREL / LUMINESCENCE / PHOTOPROTEIN / BIOLUMINESCENCE | |||||||||
| Function / homology |  Function and homology information Function and homology information | |||||||||
| Biological species |   AEQUOREA VICTORIA (jellyfish) AEQUOREA VICTORIA (jellyfish) | |||||||||
| Method | X-RAY DIFFRACTION / SAD / Resolution: 0.9 Å | |||||||||
 Authors Authors | Palm, G.J. / Schierbeek, A.J. / Kloos, M. | |||||||||
 Citation Citation | Journal: J.Am.Chem.Soc. / Year: 2010 Title: Visualizing Proton Antenna in a High-Resolution Green Fluorescent Protein Structure. Authors: Shinobu, A. / Palm, G.J. / Schierbeek, A.J. / Agmon, N. #1: Journal: Nat.Struct.Biol. / Year: 1997Title: The Structural Basis for Spectral Variations in Green Fluorescent Protein. Authors: Palm, G.J. / Zdanov, A. / Gaitanaris, G.A. / Stauber, R. / Pavlakis, G.N. / Wlodawer, A. | |||||||||
| History |
| |||||||||
| Remark 700 | SHEET DETERMINATION METHOD: DSSP THE SHEETS PRESENTED AS "AA" IN EACH CHAIN ON SHEET RECORDS BELOW ... SHEET DETERMINATION METHOD: DSSP THE SHEETS PRESENTED AS "AA" IN EACH CHAIN ON SHEET RECORDS BELOW IS ACTUALLY AN 11-STRANDED BARREL THIS IS REPRESENTED BY A 12-STRANDED SHEET IN WHICH THE FIRST AND LAST STRANDS ARE IDENTICAL. |
- Structure visualization
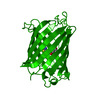
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2wur.cif.gz | 170.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2wur.ent.gz | 136.6 KB | Display | PDB format |
| PDBx/mmJSON format | 2wur.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/wu/2wurftp://data.pdbj.org/pub/pdb/validation_reports/wu/2wur | HTTPS FTP |
|---|
-Related structure data
| Related structure data | |
|---|---|
| Similar structure data |












































































-Links
 PDBj
PDBj







- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 26870.207 Da / Num. of mol.: 1 / Mutation: YES Source method: isolated from a genetically manipulated source Details: CHROMOPHORE FORMED BY RESIDUES 65-67 / Source: (gene. exp.) AEQUOREA VICTORIA (jellyfish) / Plasmid: PET11A / Production host:  ESCHERICHIA COLI (E. coli) / Strain (production host): BL21 / References: UniProt: P42212 ESCHERICHIA COLI (E. coli) / Strain (production host): BL21 / References: UniProt: P42212 | ||||||
|---|---|---|---|---|---|---|---|
| #2: Chemical | ChemComp-IPA / Isopropyl alcohol  Mass: 60.095 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O / Comment: alkaloid*YM Mass: 60.095 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O / Comment: alkaloid*YM | ||||||
| #3: Chemical | ChemComp-EOH / Ethanol  Mass: 46.068 Da / Num. of mol.: 7 / Source method: obtained synthetically / Formula: C2H6O Mass: 46.068 Da / Num. of mol.: 7 / Source method: obtained synthetically / Formula: C2H6O#4: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 318 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 318 / Source method: isolated from a natural source / Formula: H2OCompound details | ENGINEERED RESIDUE IN CHAIN A, PHE 64 TO LEU ENGINEERED RESIDUE IN CHAIN A, GLN 80 TO ARG ...ENGINEERED | Nonpolymer details | GYS: THE CHROMOPHORE (GYS) IS FORMED FROM SER 65 - TYR 66 - GLY 67 BY CYCLIZATION OF THE ...GYS: THE CHROMOPHOR | |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2 Å3/Da / Density % sol: 39 % / Description: SAD WAS BASED ON THE SULFUR SIGNAL UP TO 1.5 A |
|---|---|
| Crystal grow | Method: vapor diffusion, hanging drop / pH: 8 Details: HANGING DROP VAPOR DIFFUSION: 2 UL PROTEIN (10 MG/ML IN 20 MM TRIS, PH 8.0) PLUS 2 UL RESERVOIR (40% ETHANOL, 10 % DIOXANE) |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: BRUKER AXS MICROSTAR / Wavelength: 1.5418 |
| Detector | Type: BRUKER SMART 6000 / Detector: CCD / Date: Aug 20, 2004 / Details: MONTEL MULTILAYER OPTIC |
| Radiation | Monochromator: MIRROR / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 0.9→68 Å / Num. obs: 155426 / % possible obs: 91.4 % / Observed criterion σ(I): 0 / Redundancy: 19.7 % / Rmerge(I) obs: 0.08 / Net I/σ(I): 20.35 |
| Reflection shell | Resolution: 0.9→1 Å / Redundancy: 9.2 % / Rmerge(I) obs: 0.49 / Mean I/σ(I) obs: 3 / % possible all: 90.3 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: SAD Starting model: NONE Resolution: 0.9→6 Å / Num. parameters: 20655 / Num. restraintsaints: 26011 / Cross valid method: THROUGHOUT / σ(F): 0 / Stereochemistry target values: ENGH AND HUBER Details: ANISOTROPIC REFINEMENT REDUCED FREE R (NO CUTOFF) BY 0.03.THERE ARE MANY WEAK REFLECTIONS WHICH ARE REMOVED IN THE CALCULATION OF REFLECTIONS WITH F>4SIG(F). THERE ARE MORE REFLECTIONS WITH ...Details: ANISOTROPIC REFINEMENT REDUCED FREE R (NO CUTOFF) BY 0.03.THERE ARE MANY WEAK REFLECTIONS WHICH ARE REMOVED IN THE CALCULATION OF REFLECTIONS WITH F>4SIG(F). THERE ARE MORE REFLECTIONS WITH F>0SIG(F) NUMBERING OF THE WATER MOLECULES IS SHIFTED BY 2000 COMPARED TO THE NUMBERS USED IN THE REFERENCE LISTED IN JRNL.
| |||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: MOEWS & KRETSINGER | |||||||||||||||||||||||||||||||||
| Refine analyze | Num. disordered residues: 29 / Occupancy sum hydrogen: 1814.6 / Occupancy sum non hydrogen: 2160.4 | |||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 0.9→6 Å
| |||||||||||||||||||||||||||||||||
| Refine LS restraints |
|
