 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1a28 | ||||||
|---|---|---|---|---|---|---|---|
| Title | HORMONE-BOUND HUMAN PROGESTERONE RECEPTOR LIGAND-BINDING DOMAIN | ||||||
 Components Components | PROGESTERONE RECEPTOR | ||||||
 Keywords Keywords | PROGESTERONE RECEPTOR / STEROID RECEPTOR / NUCLEAR RECEPTOR / TRANSCRIPTION REGULATION | ||||||
| Function / homology |  Function and homology information Function and homology informationglandular epithelial cell maturation / tertiary branching involved in mammary gland duct morphogenesis / ovulation from ovarian follicle / paracrine signaling / regulation of epithelial cell proliferation / nuclear steroid receptor activity / lung alveolus development / progesterone receptor signaling pathway / estrogen response element binding / intracellular steroid hormone receptor signaling pathway ...glandular epithelial cell maturation / tertiary branching involved in mammary gland duct morphogenesis / ovulation from ovarian follicle / paracrine signaling / regulation of epithelial cell proliferation / nuclear steroid receptor activity / lung alveolus development / progesterone receptor signaling pathway / estrogen response element binding / intracellular steroid hormone receptor signaling pathway / Nuclear signaling by ERBB4 / HSP90 chaperone cycle for steroid hormone receptors (SHR) in the presence of ligand / steroid binding / G protein-coupled receptor activity / SUMOylation of intracellular receptors / transcription coactivator binding / Nuclear Receptor transcription pathway / nuclear receptor activity / cell-cell signaling / ATPase binding / DNA-binding transcription activator activity, RNA polymerase II-specific / Estrogen-dependent gene expression / mitochondrial outer membrane / nucleic acid binding / DNA-binding transcription factor activity, RNA polymerase II-specific / RNA polymerase II cis-regulatory region sequence-specific DNA binding / negative regulation of gene expression / signaling receptor binding / chromatin / positive regulation of gene expression / regulation of transcription by RNA polymerase II / enzyme binding / signal transduction / positive regulation of transcription by RNA polymerase II / DNA binding / zinc ion binding / nucleoplasm / identical protein binding / plasma membrane / cytosolSimilarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MAD, MIR / Resolution: 1.8 Å | ||||||
 Authors Authors | Sigler, P.B. / Williams, S.P. | ||||||
 Citation Citation | Journal: Nature / Year: 1998 Title: Atomic structure of progesterone complexed with its receptor. Authors: Williams, S.P. / Sigler, P.B. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1a28.cif.gz | 114.9 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1a28.ent.gz | 89.1 KB | Display | PDB format |
| PDBx/mmJSON format | 1a28.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/a2/1a28ftp://data.pdbj.org/pub/pdb/validation_reports/a2/1a28 | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|




















-Links
 PDBj
PDBj




- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
| ||||||||
| Noncrystallographic symmetry (NCS) | NCS oper: (Code: given Matrix: (0.536461, -0.825673, 0.174566), Vector : |
-Components
| #1: Protein | Mass: 29554.633 Da / Num. of mol.: 2 / Fragment: LIGAND BINDING DOMAIN Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Description: BREAST CANCER / Cell line: T47-D / Gene: PGR / Plasmid: PPR677-933 / Species (production host): Escherichia coli / Gene (production host): PGR / Production host:  Escherichia coli BL21 (bacteria) / Strain (production host): BL21 / References: UniProt: P06401 Escherichia coli BL21 (bacteria) / Strain (production host): BL21 / References: UniProt: P06401#2: Chemical | Progesterone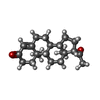  Mass: 314.462 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C21H30O2 / Comment: hormone*YM Mass: 314.462 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C21H30O2 / Comment: hormone*YM#3: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 180 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 180 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.29 Å3/Da / Density % sol: 45.7 % | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 6.5 Details: PROTEIN WAS CRYSTALLIZED FROM 2% PEG 4000, 50 MM PIPES PH 6.5, 350 MM LI2SO4, THEN STABILIZED IN THE SAME BUFFER WITH 21% ETHYLENE GLYCOL | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 4 ℃ / pH: 7.5 / Method: vapor diffusion | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: NSLS  / Beamline: X25 / Wavelength: 1.1 / Beamline: X25 / Wavelength: 1.1 |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE / Date: Sep 12, 1997 / Details: SILICON CRYSTAL |
| Radiation | Monochromator: SI(111) / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.1 Å / Relative weight: 1 |
| Reflection | Resolution: 1.8→40 Å / Num. obs: 42976 / % possible obs: 93.2 % / Observed criterion σ(I): 2 / Redundancy: 3.2 % / Biso Wilson estimate: 27 Å2 / Rmerge(I) obs: 0.055 / Rsym value: 0.076 / Net I/σ(I): 18.3 |
| Reflection shell | Resolution: 1.8→1.86 Å / Redundancy: 2.4 % / Rmerge(I) obs: 0.055 / Mean I/σ(I) obs: 3.4 / Rsym value: 0.319 / % possible all: 95.9 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MAD, MIR / Resolution: 1.8→40 Å / Data cutoff high absF: 10000 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 54.73 Å2 / ksol: 0.377 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 32.8 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.8→40 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.8→1.86 Å / Total num. of bins used: 10
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: CNS / Version: 0.1 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Rfactor Rfree: 0.2279 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Rfactor Rfree: 0.252 / Rfactor Rwork: 0.208 |
