 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-4416 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | F97L counting mode | |||||||||
 Map data Map data | ||||||||||
 Sample Sample |
| |||||||||
| Biological species |    Hepatitis B virus Hepatitis B virus | |||||||||
| Method | single particle reconstruction / cryo EM / Resolution: 2.36 Å | |||||||||
 Authors Authors | Song B / Flegler V / Makbul C / Rasmussen T / Bottcher B | |||||||||
 Citation Citation | Journal: Ultramicroscopy / Year: 2019 Title: Capabilities of the Falcon III detector for single-particle structure determination. Authors: Boyuan Song / Julian Lenhart / Vanessa Judith Flegler / Cihan Makbul / Tim Rasmussen / Bettina Böttcher /  Abstract: Direct electron detectors are an essential asset for the resolution revolution in electron cryo microscopy of biological objects. The direct detectors provide two modes of data acquisition; the ...Direct electron detectors are an essential asset for the resolution revolution in electron cryo microscopy of biological objects. The direct detectors provide two modes of data acquisition; the counting mode in which single electrons are counted, and the integrating mode in which the signal that arises from the incident electrons is integrated. While counting mode leads to far higher detective quantum efficiency at all spatial frequencies, the integrating mode enables faster data acquisition at higher exposure rates. For optimal throughput at best possible resolution it is important to understand when the better performance in counting mode becomes essential for solving a structure and when the lower detective quantum efficiency in integrating mode can be compensated by increasing the number of particles in the data set. Here, we provide a case study of the Falcon III camera, which has counting mode capability at exposure rates of <0.9 e/Px² and integrating mode capability at exposure rates above 10 e/Px². We found that counting mode gives better resolution for medium sized complexes such as the β-galactosidase (465 kDa) (2.2 Å, 97% of Nyquist vs. 2.4 Å, 89% of Nyquist) with data sets of similar size. However, for larger particles such as Hepatitis B virus capsid like particles (4.8 MDa) we did not find any resolution gain in counting mode. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
 Movie viewer Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |

- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_4416.map.gz | 227 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-4416-v30.xmlemd-4416.xml | 18.8 KB 18.8 KB | Display Display | EMDB header |
| FSC (resolution estimation) | emd_4416_fsc.xml | 27.9 KB | Display | FSC data file |
| Images |  emd_4416.png emd_4416.png | 122 KB | ||
| Others | emd_4416_half_map_1.map.gzemd_4416_half_map_2.map.gz | 223.1 MB 223.1 MB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-4416ftp://ftp.pdbj.org/pub/emdb/structures/EMD-4416 http://ftp.pdbj.org/pub/emdb/structures/EMD-4416ftp://ftp.pdbj.org/pub/emdb/structures/EMD-4416 | HTTPS FTP |
-Related structure data














-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|
-Map
| File | Download / File: emd_4416.map.gz / Format: CCP4 / Size: 244.1 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Voxel size | X=Y=Z: 1.0635 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
-Supplemental data
-Half map: #2
| File | emd_4416_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
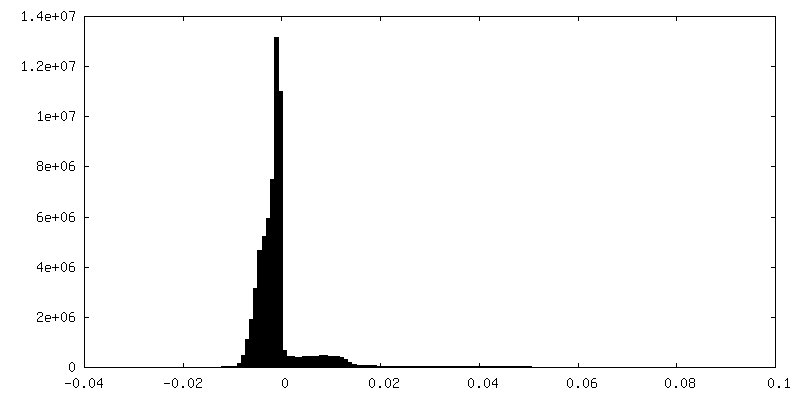
| Density Histograms |
 Z
Z Y
Y X
X




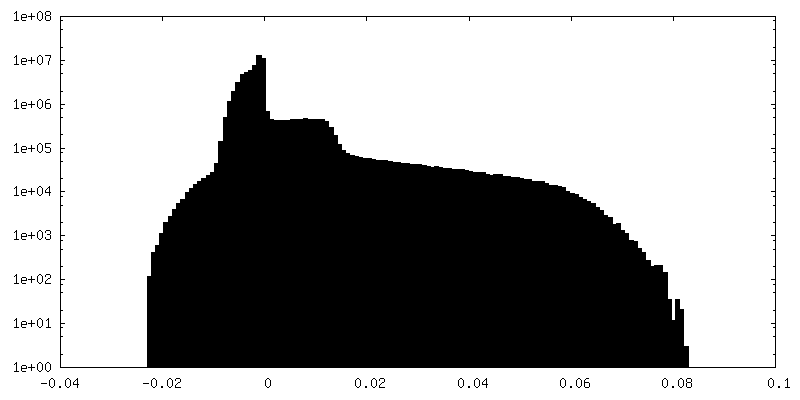
-Half map: #1
| File | emd_4416_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
| Density Histograms |



- Sample components
Sample components
-Entire : Hepatitis B virus
| Entire | Name: Hepatitis B virus |
|---|---|
| Components |
|
-Supramolecule #1: Hepatitis B virus
| Supramolecule | Name: Hepatitis B virus / type: virus / ID: 1 / Parent: 0 / Macromolecule list: all / Details: core protein was expressed in E.coli / NCBI-ID: 10407 / Sci species name: Hepatitis B virus / Virus type: VIRUS-LIKE PARTICLE / Virus isolate: OTHER / Virus enveloped: No / Virus empty: No |
|---|---|
| Host (natural) | Organism:  Homo sapiens (human) Homo sapiens (human) |
| Host system | Organism:  Escherichia coli (E. coli) Escherichia coli (E. coli) |
| Molecular weight | Theoretical: 4.8 MDa |
| Virus shell | Shell ID: 1 / Diameter: 340.0 Å / T number (triangulation number): 4 |
-Macromolecule #1: Hepatitis B virus core protein
| Macromolecule | Name: Hepatitis B virus core protein / type: protein_or_peptide / ID: 1 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: Hepatitis B virus |
| Recombinant expression | Organism: Escherichia coli (E. coli) |
| Sequence | String: MDIDPYKEFG ATVELLSFLP SDFFPSVRDL LDTASALYRE ALESPEHCSP HHTALRQAIL CWGELMTLAT WVGVNLEDP ASRDLVVSYV NTNMGLKLRQ LLWFHISCLT FGRETVIEYL VSFGVWIRTP PAYRPPNAPI L STLPETTV VRRRGRSPRK RTPSPRKRRS QSPRRRRSQS RESQC |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 4 mg/mL |
|---|---|
| Buffer | pH: 7.5 / Details: 25 mM Tris |
| Grid | Model: Quantifoil, UltrAuFoil, R1.2/1.3 / Material: GOLD / Mesh: 300 / Support film - topology: HOLEY / Support film - Film thickness: 50.0 nm / Pretreatment - Type: GLOW DISCHARGE / Pretreatment - Atmosphere: AIR |
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 100 % / Chamber temperature: 277 K / Instrument: FEI VITROBOT MARK IV Details: Quantifoil 300 mesh gold ultrafoil grids R 1.2/1.3 were used (Quantifoil Micro Tools GmbH, Jena, Germany). Grids were glow discharged in air at a pressure of 2.2x10-2 Torr for 2 min at ...Details: Quantifoil 300 mesh gold ultrafoil grids R 1.2/1.3 were used (Quantifoil Micro Tools GmbH, Jena, Germany). Grids were glow discharged in air at a pressure of 2.2x10-2 Torr for 2 min at medium power with a Harrick Plasma cleaner (PDC-002). Subsequently, 3-4 ul of the sample was pipetted onto the glow discharged grids and plunge frozen in liquid Ethane with a Vitrobot IV (FEI). The sample chamber of the Vitrobot was maintained at 4C and 100% humidity. Wait time between sample application and blotting was 45 s, the drain time 0 s, the blot time 3 s and the blot force 0.. |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Electron beam | Acceleration voltage: 300 kV / Electron source: FIELD EMISSION GUN |
| Electron optics | C2 aperture diameter: 70.0 µm / Calibrated defocus max: 2.57 µm / Calibrated defocus min: 0.68 µm / Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELDBright-field microscopy / Cs: 2.7 mm / Nominal defocus max: 2.0 µm / Nominal defocus min: 1.0 µm / Nominal magnification: 75000 |
| Sample stage | Specimen holder model: FEI TITAN KRIOS AUTOGRID HOLDER / Cooling holder cryogen: NITROGEN |
| Temperature | Min: 79.0 K / Max: 80.0 K |
| Details | data has been recorded in counting mode 40 frames per movie |
| Image recording | Film or detector model: FEI FALCON III (4k x 4k) / Detector mode: COUNTING / Digitization - Dimensions - Width: 4096 pixel / Digitization - Dimensions - Height: 4096 pixel / Number grids imaged: 1 / Number real images: 1616 / Average exposure time: 75.0 sec. / Average electron dose: 40.0 e/Å2 Details: images were collected in movie mode (31 frames in total) |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
-Image processing
| Particle selection | Number selected: 88711 |
|---|---|
| CTF correction | Software - Name: RELION |
| Startup model | Type of model: EMDB MAP EMDB ID: Details: previously determined |
| Initial angle assignment | Type: MAXIMUM LIKELIHOOD / Software - Name: RELION (ver. 3.0) |
| Final 3D classification | Number classes: 3 / Software - Name: RELION (ver. 3.0) |
| Final angle assignment | Type: MAXIMUM LIKELIHOOD / Software - Name: RELION (ver. 3.0) |
| Final reconstruction | Number classes used: 1 / Applied symmetry - Point group: I (icosahedral) / Algorithm: FOURIER SPACE / Resolution.type: BY AUTHOR / Resolution: 2.36 Å / Resolution method: FSC 0.143 CUT-OFF / Software - Name: RELION (ver. 3.0) / Number images used: 45444 |
| Details | Movies were motion corrected and exposure weighted with motioncor2. The defocus was determined for the motion corrected and exposure weighted averages with ctffind4 |
| FSC plot (resolution estimation) | 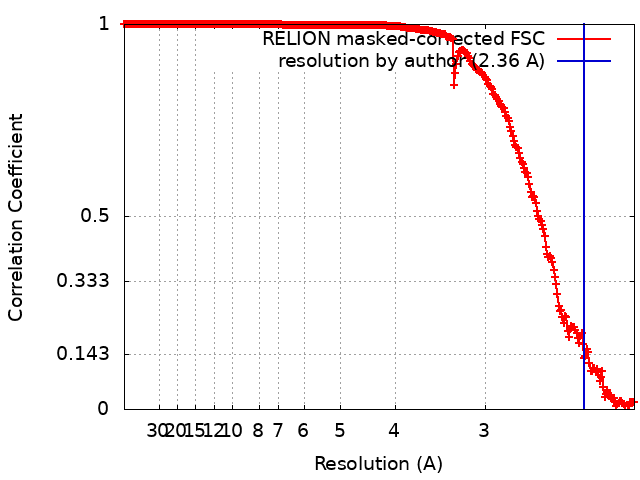 |

-Atomic model buiding 1
| Refinement | Space: RECIPROCAL / Protocol: AB INITIO MODEL / Overall B value: 75 |
|---|
