 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-5w0u: Crystal structure of MBP fused activation-induced cytidine deamin... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 5w0u | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of MBP fused activation-induced cytidine deaminase (AID) in complex with dCMP | ||||||
 Components Components |
| ||||||
 Keywords Keywords | HYDROLASE/DNA / Class switch recombination / Cytidine deaminase / HYDROLASE-DNA complex | ||||||
| Function / homology |  Function and homology information Function and homology informationsomatic diversification of immunoglobulins / regulation of nuclear cell cycle DNA replication / single-stranded DNA cytosine deaminase / negative regulation of single stranded viral RNA replication via double stranded DNA intermediate / DNA cytosine deamination / cytidine to uridine editing / cytidine deaminase activity / positive regulation of gene expression via chromosomal CpG island demethylation / isotype switching / carbohydrate transmembrane transporter activity ...somatic diversification of immunoglobulins / regulation of nuclear cell cycle DNA replication / single-stranded DNA cytosine deaminase / negative regulation of single stranded viral RNA replication via double stranded DNA intermediate / DNA cytosine deamination / cytidine to uridine editing / cytidine deaminase activity / positive regulation of gene expression via chromosomal CpG island demethylation / isotype switching / carbohydrate transmembrane transporter activity / maltose binding / maltose transport / maltodextrin transmembrane transport / somatic hypermutation of immunoglobulin genes / ATP-binding cassette (ABC) transporter complex, substrate-binding subunit-containing / B cell differentiation / Chromatin modifications during the maternal to zygotic transition (MZT) / P-body / mRNA processing / outer membrane-bounded periplasmic space / cellular response to lipopolysaccharide / defense response to virus / defense response to bacterium / ubiquitin protein ligase binding / protein-containing complex / RNA binding / zinc ion binding / identical protein binding / nucleus / cytoplasm / cytosol Similarity search - Function | ||||||
| Biological species |   Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.9 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.9 Å | ||||||
 Authors Authors | Qiao, Q. / Wang, L. / Wu, H. | ||||||
 Citation Citation | Journal: Mol. Cell / Year: 2017 Title: AID Recognizes Structured DNA for Class Switch Recombination. Authors: Qiao, Q. / Wang, L. / Meng, F.L. / Hwang, J.K. / Alt, F.W. / Wu, H. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 5w0u.cif.gz | 242.4 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb5w0u.ent.gz | 190 KB | Display | PDB format |
| PDBx/mmJSON format | 5w0u.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/w0/5w0uftp://data.pdbj.org/pub/pdb/validation_reports/w0/5w0u | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  5w0rSC  5w0zC  5w1cC S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj









































- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
|
-Components
-Protein , 1 types, 2 molecules BA
| #1: Protein | Mass: 61659.883 Da / Num. of mol.: 2 / Fragment: UNP residues 27-392,UNP residues 13-181 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human)Gene: malE, Z5632, ECs5017, AICDA, AID / Production host:   Spodoptera frugiperda (fall armyworm) Spodoptera frugiperda (fall armyworm)References: UniProt: P0AEY0, UniProt: Q9GZX7, single-stranded DNA cytosine deaminase |
|---|
-DNA chain , 2 types, 2 molecules DG
| #2: DNA chain | Mass: 3687.417 Da / Num. of mol.: 1 / Source method: obtained synthetically / Source: (synth.) Homo sapiens (human) |
|---|---|
| #3: DNA chain | Mass: 3638.379 Da / Num. of mol.: 1 / Source method: obtained synthetically / Source: (synth.) Homo sapiens (human) |
-Non-polymers , 5 types, 9 molecules 
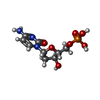



| #4: Chemical |  Mass: 65.409 Da / Num. of mol.: 2 Mass: 65.409 Da / Num. of mol.: 2Source method: isolated from a genetically manipulated source Formula: Zn #5: Chemical |  Mass: 307.197 Da / Num. of mol.: 2 Mass: 307.197 Da / Num. of mol.: 2Source method: isolated from a genetically manipulated source Formula: C9H14N3O7P #6: Chemical |  Mass: 40.078 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Ca Mass: 40.078 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Ca#7: Chemical | ChemComp-GOL / |  Mass: 92.094 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 92.094 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O3#8: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 2 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.95 Å3/Da / Density % sol: 58.26 % |
|---|---|
| Crystal grow | Temperature: 289 K / Method: vapor diffusion, hanging drop / pH: 6.2 Details: 0.1 M MES at pH 6.2, 3%PEG3350, 10 mM CaCl2, 20mM dCMP |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS  / Beamline: 24-ID-C / Wavelength: 0.9791 Å / Beamline: 24-ID-C / Wavelength: 0.9791 Å |
| Detector | Type: DECTRIS PILATUS 6M-F / Detector: PIXEL / Date: Apr 20, 2016 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9791 Å / Relative weight: 1 |
| Reflection | Resolution: 2.9→153.935 Å / Num. obs: 36653 / % possible obs: 98.3 % / Redundancy: 9.8 % / Rmerge(I) obs: 0.094 / Net I/σ(I): 12.1 |
| Reflection shell | Resolution: 2.94→3.07 Å / Redundancy: 10.2 % / Rmerge(I) obs: 0.656 / Mean I/σ(I) obs: 2.8 / Num. unique obs: 3868 / CC1/2: 0.974 / Rpim(I) all: 0.313 / % possible all: 99.7 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 5W0R Resolution: 2.9→153.935 Å / SU ML: 0.39 / Cross valid method: FREE R-VALUE / σ(F): 1.34 / Phase error: 39.63 / Stereochemistry target values: ML
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.9→153.935 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION / Rfactor Rfree error: 0 / Total num. of bins used: 10
|
