 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 5uat | ||||||
|---|---|---|---|---|---|---|---|
| Title | Structure of human PYCR-1 complexed with NADPH | ||||||
 Components Components | Pyrroline-5-carboxylate reductase 1, mitochondrial | ||||||
 Keywords Keywords | OXIDOREDUCTASE / AMINO-ACID BIOSYNTHESIS / PROLINE BIOSYNTHESIS | ||||||
| Function / homology |  Function and homology information Function and homology informationpyrroline-5-carboxylate reductase complex / pyrroline-5-carboxylate reductase / pyrroline-5-carboxylate reductase activity / Glutamate and glutamine metabolism / L-proline biosynthetic process / negative regulation of oxidative stress-induced neuron intrinsic apoptotic signaling pathway / regulation of mitochondrial membrane potential / cellular response to oxidative stress / mitochondrial matrix / mitochondrion / identical protein binding Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.92 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.92 Å | ||||||
 Authors Authors | Tanner, J.J. | ||||||
| Funding support |  United States, 1items United States, 1items
| ||||||
 Citation Citation | Journal: J. Biol. Chem. / Year: 2017 Title: Resolving the cofactor-binding site in the proline biosynthetic enzyme human pyrroline-5-carboxylate reductase 1. Authors: Christensen, E.M. / Patel, S.M. / Korasick, D.A. / Campbell, A.C. / Krause, K.L. / Becker, D.F. / Tanner, J.J. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 5uat.cif.gz | 512.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb5uat.ent.gz | 420.2 KB | Display | PDB format |
| PDBx/mmJSON format | 5uat.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ua/5uatftp://data.pdbj.org/pub/pdb/validation_reports/ua/5uat | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  5uauC  5uavC  5uawC  5uaxC  2izzS C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj




- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| 3 | 
| ||||||||
| 4 | 
| ||||||||
| Unit cell |
| ||||||||
| Details | The authors state that based on analytical ultracentrifugation sedimentation velocity experiments, the protein is primarily a decamer at 6 mg/mL (180 micro molar based on monomer molecular weight). Lower molecular weight species are also observed at a protein concentration of 0.8 mg/mL. |
-Components
| #1: Protein | Mass: 34043.156 Da / Num. of mol.: 5 / Fragment: residues 1-300 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: PYCR1 / Production host:  References: UniProt: P32322, pyrroline-5-carboxylate reductase #2: Chemical | ChemComp-SO4 /   Mass: 96.063 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: SO4#3: Chemical | 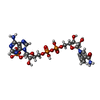  Mass: 745.421 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C21H30N7O17P3 Mass: 745.421 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C21H30N7O17P3#4: Chemical | ChemComp-PEG / |   Mass: 106.120 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H10O3 Mass: 106.120 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H10O3#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 557 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 557 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.42 Å3/Da / Density % sol: 49.2 % |
|---|---|
| Crystal grow | Temperature: 295 K / Method: vapor diffusion, sitting drop / pH: 7.5 Details: 300 mM Na2SO4, 16-18% (w/v) polyethylene glycol (PEG) 3350, and 0.1 M HEPES at pH 7.5. The crystal was soaked in the cryobuffer containing 100 mM NADPH |
-Data collection
| Diffraction | Mean temperature: 100 K | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ALS / Beamline: 4.2.2 / Wavelength: 1 Å | ||||||||||||||||||
| Detector | Type: RDI CMOS_8M / Detector: CMOS / Date: Oct 18, 2015 | ||||||||||||||||||
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | ||||||||||||||||||
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 | ||||||||||||||||||
| Reflection | Resolution: 1.92→64.24 Å / Num. obs: 126545 / % possible obs: 100 % / Redundancy: 6.9 % / Biso Wilson estimate: 24.25 Å2 / CC1/2: 0.999 / Rmerge(I) obs: 0.094 / Rpim(I) all: 0.038 / Rrim(I) all: 0.101 / Net I/σ(I): 14.9 / Num. measured all: 879308 / Scaling rejects: 80 | ||||||||||||||||||
| Reflection shell |
|
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 2IZZ Resolution: 1.92→64.235 Å / SU ML: 0.19 / Cross valid method: FREE R-VALUE / σ(F): 1.35 / Phase error: 20.7
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 101.8 Å2 / Biso mean: 32.3083 Å2 / Biso min: 10.21 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 1.92→64.235 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION / Rfactor Rfree error: 0 / Total num. of bins used: 30 / % reflection obs: 100 %
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
