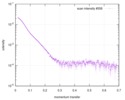
Journal: Angew Chem Int Ed Engl / Year: 2017 Title: Segmental, Domain-Selective Perdeuteration and Small-Angle Neutron Scattering for Structural Analysis of Multi-Domain Proteins. Authors: Miriam Sonntag / Pravin Kumar Ankush Jagtap / Bernd Simon / Marie-Sousai Appavou / Arie Geerlof / Ralf Stehle / Frank Gabel / Janosch Hennig / Michael Sattler / Abstract: Multi-domain proteins play critical roles in fine-tuning essential processes in cellular signaling and gene regulation. Typically, multiple globular domains that are connected by flexible linkers ...Multi-domain proteins play critical roles in fine-tuning essential processes in cellular signaling and gene regulation. Typically, multiple globular domains that are connected by flexible linkers undergo dynamic rearrangements upon binding to protein, DNA or RNA ligands. RNA binding proteins (RBPs) represent an important class of multi-domain proteins, which regulate gene expression by recognizing linear or structured RNA sequence motifs. Here, we employ segmental perdeuteration of the three RNA recognition motif (RRM) domains in the RBP TIA-1 using Sortase A mediated protein ligation. We show that domain-selective perdeuteration combined with contrast-matched small-angle neutron scattering (SANS), SAXS and computational modeling provides valuable information to precisely define relative domain arrangements. The approach is generally applicable to study conformational arrangements of individual domains in multi-domain proteins and changes induced by ligand binding.
Resolution: 2.97→35.02 Å / Cor.coef. Fo:Fc: 0.919 / Cor.coef. Fo:Fc free: 0.909 / SU B: 31.878 / SU ML: 0.526 / Cross valid method: THROUGHOUT / ESU R Free: 0.557 / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS
Rfactor
Num. reflection
% reflection
Selection details
Rfree
0.3116
98
5 %
RANDOM
Rwork
0.25095
-
-
-
obs
0.25387
1859
98.14 %
-
Solvent computation
Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information Homo sapiens (human)
Homo sapiens (human) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation

 Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads















 PDBj
PDBj
 Assembly
Assembly

 Sample preparation
Sample preparation Processing
Processing