 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-5j3s: Crystal structure of the catalytic domain of human tyrosyl DNA ph... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 5j3s | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of the catalytic domain of human tyrosyl DNA phosphodiesterase 2 in complex with a small molecule inhibitor | ||||||
 Components Components | Tyrosyl-DNA phosphodiesterase 2 | ||||||
 Keywords Keywords | HYDROLASE / tyrosyl DNA phosphodiesterase 2 catalytic domain | ||||||
| Function / homology |  Function and homology information Function and homology informationtyrosyl-RNA phosphodiesterase activity / 5'-tyrosyl-DNA phosphodiesterase activity / nuclease activity / Hydrolases; Acting on ester bonds; Phosphoric-diester hydrolases / aggresome / neuron development / Nonhomologous End-Joining (NHEJ) / PML body / transcription corepressor activity / double-strand break repair ...tyrosyl-RNA phosphodiesterase activity / 5'-tyrosyl-DNA phosphodiesterase activity / nuclease activity / Hydrolases; Acting on ester bonds; Phosphoric-diester hydrolases / aggresome / neuron development / Nonhomologous End-Joining (NHEJ) / PML body / transcription corepressor activity / double-strand break repair / manganese ion binding / single-stranded DNA binding / nuclear body / cell surface receptor signaling pathway / nucleolus / magnesium ion binding / nucleoplasm / nucleus / cytoplasm Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 3.4 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 3.4 Å | ||||||
 Authors Authors | Hornyak, P. / Pearl, L.H. / Caldecott, K.W. / Oliver, A.W. | ||||||
 Citation Citation | Journal: Biochem.J. / Year: 2016 Title: Mode of action of DNA-competitive small molecule inhibitors of tyrosyl DNA phosphodiesterase 2. Authors: Hornyak, P. / Askwith, T. / Walker, S. / Komulainen, E. / Paradowski, M. / Pennicott, L.E. / Bartlett, E.J. / Brissett, N.C. / Raoof, A. / Watson, M. / Jordan, A.M. / Ogilvie, D.J. / Ward, S. ...Authors: Hornyak, P. / Askwith, T. / Walker, S. / Komulainen, E. / Paradowski, M. / Pennicott, L.E. / Bartlett, E.J. / Brissett, N.C. / Raoof, A. / Watson, M. / Jordan, A.M. / Ogilvie, D.J. / Ward, S.E. / Atack, J.R. / Pearl, L.H. / Caldecott, K.W. / Oliver, A.W. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 5j3s.cif.gz | 107.3 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb5j3s.ent.gz | 82.1 KB | Display | PDB format |
| PDBx/mmJSON format | 5j3s.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/j3/5j3sftp://data.pdbj.org/pub/pdb/validation_reports/j3/5j3s | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  5j3pSC  5j3zC  5j42C S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj





- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 28824.314 Da / Num. of mol.: 1 / Mutation: C273S Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: TDP2, EAP2, TTRAP, AD-022 / Production host:  References: UniProt: O95551, Hydrolases; Acting on ester bonds; Phosphoric-diester hydrolases |
|---|---|
| #2: Chemical | ChemComp-6FQ / 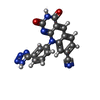  Mass: 382.335 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C19H10N8O2 Mass: 382.335 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C19H10N8O2 |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.61 Å3/Da / Density % sol: 52.84 % |
|---|---|
| Crystal grow | Temperature: 273 K / Method: vapor diffusion, hanging drop Details: 1.2M D/L-Malic acid pH7.0, 0.1M Bis-Tris propane pH 7.0, 3% v/v DMSO |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: Diamond  / Beamline: I04 / Wavelength: 0.9795 Å / Beamline: I04 / Wavelength: 0.9795 Å |
| Detector | Type: DECTRIS PILATUS 6M-F / Detector: PIXEL / Date: Feb 24, 2013 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9795 Å / Relative weight: 1 |
| Reflection | Resolution: 3.4→45.22 Å / Num. obs: 7545 / % possible obs: 99.9 % / Redundancy: 6.3 % / CC1/2: 0.99 / Rmerge(I) obs: 0.2423 / Net I/σ(I): 6.4 |
| Reflection shell | Resolution: 3.4→3.67 Å / Redundancy: 6.8 % / Rmerge(I) obs: 2.025 / Mean I/σ(I) obs: 1.1 / CC1/2: 0.54 / % possible all: 100 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 5J3P Resolution: 3.4→45.218 Å / SU ML: 0.48 / Cross valid method: FREE R-VALUE / σ(F): 0.01 / Phase error: 32.22
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 3.4→45.218 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
