| Entry | Database: PDB / ID: 5g6v
|
|---|
| Title | Crystal structure of the PCTAIRE1 kinase in complex with inhibitor |
|---|
 Components Components | CYCLIN-DEPENDENT KINASE 16 |
|---|
 Keywords Keywords | TRANSFERASE |
|---|
| Function / homology |  Function and homology information Function and homology information
growth hormone secretion / regulation of cell cycle phase transition / regulation of insulin secretion involved in cellular response to glucose stimulus / exocytosis / cyclin-dependent kinase / cyclin-dependent protein serine/threonine kinase activity / cyclin-dependent protein kinase holoenzyme complex / positive regulation of autophagy / neuron projection development / cytoplasmic side of plasma membrane ...growth hormone secretion / regulation of cell cycle phase transition / regulation of insulin secretion involved in cellular response to glucose stimulus / exocytosis / cyclin-dependent kinase / cyclin-dependent protein serine/threonine kinase activity / cyclin-dependent protein kinase holoenzyme complex / positive regulation of autophagy / neuron projection development / cytoplasmic side of plasma membrane / synaptic vesicle / spermatogenesis / protein phosphorylation / neuron projection / protein serine kinase activity / protein serine/threonine kinase activity / ATP binding / nucleus / plasma membrane / cytosol / cytoplasmSimilarity search - Function : / Phosphorylase Kinase; domain 1 / Phosphorylase Kinase; domain 1 / Transferase(Phosphotransferase) domain 1 / Transferase(Phosphotransferase); domain 1 / Serine/threonine-protein kinase, active site / Serine/Threonine protein kinases active-site signature. / Protein kinase domain / Serine/Threonine protein kinases, catalytic domain / Protein kinase, ATP binding site ...: / Phosphorylase Kinase; domain 1 / Phosphorylase Kinase; domain 1 / Transferase(Phosphotransferase) domain 1 / Transferase(Phosphotransferase); domain 1 / Serine/threonine-protein kinase, active site / Serine/Threonine protein kinases active-site signature. / Protein kinase domain / Serine/Threonine protein kinases, catalytic domain / Protein kinase, ATP binding site / Protein kinases ATP-binding region signature. / Protein kinase domain profile. / Protein kinase domain / Protein kinase-like domain superfamily / 2-Layer Sandwich / Orthogonal Bundle / Mainly Alpha / Alpha BetaSimilarity search - Domain/homology |
|---|
| Biological species |  HOMO SAPIENS (human) HOMO SAPIENS (human) |
|---|
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.2 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.2 Å |
|---|
 Authors Authors | Dixon-Clarke, S.E. / Galan Bartual, S. / Elkins, J. / Savitsky, P. / Kopec, J. / Mackenzie, A. / Tallant, C. / Heroven, C. / Burgess-Brown, N. / von Delft, F. ...Dixon-Clarke, S.E. / Galan Bartual, S. / Elkins, J. / Savitsky, P. / Kopec, J. / Mackenzie, A. / Tallant, C. / Heroven, C. / Burgess-Brown, N. / von Delft, F. / Arrowsmith, C.H. / Edwards, A.M. / Bountra, C. / Bullock, A. |
|---|
 Citation Citation | Journal: Biochem.J. / Year: 2017
Title: Structure and inhibitor specificity of the PCTAIRE-family kinase CDK16.
Authors: Dixon-Clarke, S.E. / Shehata, S.N. / Krojer, T. / Sharpe, T.D. / von Delft, F. / Sakamoto, K. / Bullock, A.N. |
|---|
| History | | Deposition | Aug 16, 2016 | Deposition site: PDBE / Processing site: PDBE |
|---|
| Revision 1.0 | Nov 23, 2016 | Provider: repository / Type: Initial release |
|---|
| Revision 1.1 | Aug 2, 2023 | Group: Database references / Derived calculations ...Database references / Derived calculations / Other / Refinement description
Category: citation / citation_author ...citation / citation_author / database_2 / pdbx_database_status / struct_ncs_dom_lim / struct_site
Item: _citation.country / _citation.journal_abbrev ..._citation.country / _citation.journal_abbrev / _citation.journal_id_ASTM / _citation.journal_id_CSD / _citation.journal_id_ISSN / _citation.journal_volume / _citation.page_first / _citation.page_last / _citation.pdbx_database_id_DOI / _citation.pdbx_database_id_PubMed / _citation.title / _citation.year / _database_2.pdbx_DOI / _database_2.pdbx_database_accession / _pdbx_database_status.status_code_sf / _struct_ncs_dom_lim.beg_auth_comp_id / _struct_ncs_dom_lim.beg_label_asym_id / _struct_ncs_dom_lim.beg_label_comp_id / _struct_ncs_dom_lim.beg_label_seq_id / _struct_ncs_dom_lim.end_auth_comp_id / _struct_ncs_dom_lim.end_label_asym_id / _struct_ncs_dom_lim.end_label_comp_id / _struct_ncs_dom_lim.end_label_seq_id / _struct_site.pdbx_auth_asym_id / _struct_site.pdbx_auth_comp_id / _struct_site.pdbx_auth_seq_id |
|---|
| Revision 1.2 | Jun 19, 2024 | Group: Data collection / Category: chem_comp_atom / chem_comp_bond |
|---|
|
|---|
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads










 PDBj
PDBj
 Assembly
Assembly



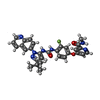
 Mass: 553.587 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C30H28FN7O3
Mass: 553.587 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C30H28FN7O3
 Mass: 62.068 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C2H6O2
Mass: 62.068 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C2H6O2 Mass: 18.015 Da / Num. of mol.: 262 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 262 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation / Beamline: I02 / Wavelength: 0.91742
/ Beamline: I02 / Wavelength: 0.91742  Processing
Processing