 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-4zyz: Human GAR transformylase in complex with GAR and (S)-2-(7-(2-Amin... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4zyz | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Human GAR transformylase in complex with GAR and (S)-2-(7-(2-Amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-6-yl)heptanamido)pentanedioic acid (AGF145) | |||||||||
 Components Components | Trifunctional purine biosynthetic protein adenosine-3 | |||||||||
 Keywords Keywords | TRANSFERASE/TRANSFERASE INHIBITOR / gar transformylase / antifolate / TRANSFERASE-TRANSFERASE INHIBITOR complex | |||||||||
| Function / homology |  Function and homology information Function and homology informationphosphoribosylformylglycinamidine cyclo-ligase / phosphoribosylformylglycinamidine cyclo-ligase activity / adenine biosynthetic process / phosphoribosylamine-glycine ligase / phosphoribosylamine-glycine ligase activity / phosphoribosylglycinamide formyltransferase 1 / purine ribonucleoside monophosphate biosynthetic process / phosphoribosylglycinamide formyltransferase activity / 'de novo' XMP biosynthetic process / brainstem development ...phosphoribosylformylglycinamidine cyclo-ligase / phosphoribosylformylglycinamidine cyclo-ligase activity / adenine biosynthetic process / phosphoribosylamine-glycine ligase / phosphoribosylamine-glycine ligase activity / phosphoribosylglycinamide formyltransferase 1 / purine ribonucleoside monophosphate biosynthetic process / phosphoribosylglycinamide formyltransferase activity / 'de novo' XMP biosynthetic process / brainstem development / Purine ribonucleoside monophosphate biosynthesis / glycine metabolic process / 'de novo' AMP biosynthetic process / purine nucleotide biosynthetic process / GMP biosynthetic process / 'de novo' IMP biosynthetic process / tetrahydrofolate biosynthetic process / cerebellum development / cerebral cortex development / extracellular exosome / ATP binding / metal ion binding / cytosol Similarity search - Function | |||||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | |||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 1.6 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 1.6 Å | |||||||||
 Authors Authors | Deis, S.M. / Dann III, C.E. | |||||||||
| Funding support |  United States, 2items United States, 2items
| |||||||||
 Citation Citation | Journal: Biochemistry / Year: 2016 Title: Structural and Enzymatic Analysis of Tumor-Targeted Antifolates That Inhibit Glycinamide Ribonucleotide Formyltransferase. Authors: Deis, S.M. / Doshi, A. / Hou, Z. / Matherly, L.H. / Gangjee, A. / Dann, C.E. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4zyz.cif.gz | 104 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4zyz.ent.gz | 77.3 KB | Display | PDB format |
| PDBx/mmJSON format | 4zyz.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Summary document | 4zyz_validation.pdf.gz | 1008.4 KB | Display | wwPDB validaton report |
|---|---|---|---|---|
| Full document | 4zyz_full_validation.pdf.gz | 1009.8 KB | Display | |
| Data in XML | 4zyz_validation.xml.gz | 12.4 KB | Display | |
| Data in CIF | 4zyz_validation.cif.gz | 17.8 KB | Display | |
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/zy/4zyzftp://data.pdbj.org/pub/pdb/validation_reports/zy/4zyz | HTTPS FTP |
-Related structure data
| Related structure data |  4zytC  4zyuC  4zyvC  4zywC  4zyxC  4zyyC  4zz0C  4x77 C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |



























-Links
 PDBj
PDBj





- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 22810.139 Da / Num. of mol.: 1 / Fragment: gar transformylase domain Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: GART, PGFT, PRGS / Plasmid: pET22B / Production host:  References: UniProt: P22102, phosphoribosylglycinamide formyltransferase 1 |
|---|---|
| #2: Chemical | ChemComp-GAR / 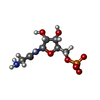  Mass: 284.160 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C7H13N2O8P Mass: 284.160 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C7H13N2O8P |
| #3: Chemical | ChemComp-3YD / (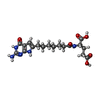  Mass: 407.421 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C18H25N5O6 Mass: 407.421 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C18H25N5O6 |
| #4: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 203 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 203 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.89 Å3/Da / Density % sol: 68.35 % |
|---|---|
| Crystal grow | Temperature: 277 K / Method: vapor diffusion, sitting drop / pH: 7.5 / Details: 18 % PEG4000, 2 % PEG400, 0.33 M NaCl |
-Data collection
| Diffraction | Mean temperature: 100 K | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ALS / Beamline: 4.2.2 / Wavelength: 1.0001 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Detector | Type: RDI CMOS_8M / Detector: CMOS / Date: Aug 24, 2014 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation | Monochromator: Rosenbaum-Rock Si(111) sagitally focused monochromator Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 1.0001 Å / Relative weight: 1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Resolution: 1.6→50 Å / Num. obs: 44634 / % possible obs: 99.5 % / Redundancy: 9.6 % / Biso Wilson estimate: 13.72 Å2 / Rmerge(I) obs: 0.063 / Rpim(I) all: 0.021 / Rrim(I) all: 0.066 / Χ2: 0.934 / Net I/av σ(I): 32.129 / Net I/σ(I): 18.8 / Num. measured all: 428756 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection shell | Diffraction-ID: 1 / Rejects: _
|
-Phasing
| Phasing | Method: molecular replacement |
|---|
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 4X77 4x77 Resolution: 1.6→39.995 Å / SU ML: 0.12 / Cross valid method: FREE R-VALUE / σ(F): 1.34 / Phase error: 16.98 / Stereochemistry target values: ML
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 78.24 Å2 / Biso mean: 21.3294 Å2 / Biso min: 7.1 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 1.6→39.995 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION / Total num. of bins used: 14
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
