 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-4zyb: High resolution structure of M23 peptidase LytM with substrate an... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4zyb | ||||||
|---|---|---|---|---|---|---|---|
| Title | High resolution structure of M23 peptidase LytM with substrate analogue | ||||||
 Components Components | Glycyl-glycine endopeptidase LytM | ||||||
 Keywords Keywords | HYDROLASE / LYTM / LYSOSTAPHIN / PEPTIDOGLYCAN AMIDASE / PEPTIDASE / TETRAGLYCINE PHOSPHINATE / TRANSITION STATE ANALOGUE / COMPLEX | ||||||
| Function / homology |  Function and homology information Function and homology informationlysostaphin / cobalt ion binding / peptide catabolic process / nickel cation binding / cell wall organization / metalloendopeptidase activity / manganese ion binding / proteolysis / extracellular region / zinc ion binding / metal ion binding Similarity search - Function | ||||||
| Biological species |  Staphylococcus aureus subsp. aureus NCTC 8325 (bacteria) Staphylococcus aureus subsp. aureus NCTC 8325 (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.5 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.5 Å | ||||||
 Authors Authors | Grabowska, M. / Jagielska, E. / Czapinska, H. / Bochtler, M. / Sabala, I. | ||||||
| Funding support |  Poland, 1items Poland, 1items
| ||||||
 Citation Citation | Journal: Sci Rep / Year: 2015 Title: High resolution structure of an M23 peptidase with a substrate analogue. Authors: Grabowska, M. / Jagielska, E. / Czapinska, H. / Bochtler, M. / Sabala, I. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4zyb.cif.gz | 264.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4zyb.ent.gz | 212.6 KB | Display | PDB format |
| PDBx/mmJSON format | 4zyb.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/zy/4zybftp://data.pdbj.org/pub/pdb/validation_reports/zy/4zyb | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  2b0pS S: Starting model for refinement |
|---|---|
| Similar structure data |



















-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| 3 | 
| ||||||||
| 4 | 
| ||||||||
| Unit cell |
|
-Components
-Protein , 1 types, 4 molecules ABCD
| #1: Protein | Mass: 14564.811 Da / Num. of mol.: 4 / Fragment: unp residues 185-316 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Staphylococcus aureus subsp. aureus NCTC 8325 (bacteria)Gene: lytM, SAS0252 / Plasmid: PLASMID / Details (production host): PET15B / Production host: References: UniProt: Q6GCJ6, UniProt: O33599*PLUS, lysostaphin |
|---|
-Non-polymers , 9 types, 889 molecules 



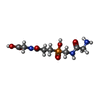



| #2: Chemical | ChemComp-ZN /  Mass: 65.409 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Zn Mass: 65.409 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Zn#3: Chemical | ChemComp-CA /  Mass: 40.078 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Ca Mass: 40.078 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Ca#4: Chemical | ChemComp-CL /  Mass: 35.453 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Cl Mass: 35.453 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Cl#5: Chemical | ChemComp-UNL / Mass: 103.120 Da / Num. of mol.: 4 / Source method: obtained synthetically #6: Chemical |  Mass: 62.068 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C2H6O2 Mass: 62.068 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C2H6O2#7: Chemical | ChemComp-4SQ /  Mass: 281.203 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C8H16N3O6P Mass: 281.203 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C8H16N3O6P#8: Chemical |  Mass: 238.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C8H18N2O4S / Comment: pH buffer*YM Mass: 238.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C8H18N2O4S / Comment: pH buffer*YM#9: Chemical | ChemComp-PEG / |  Mass: 106.120 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H10O3 Mass: 106.120 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H10O3#10: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 864 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.19 Å3/Da / Density % sol: 43.76 % |
|---|---|
| Crystal grow | Temperature: 290 K / Method: vapor diffusion, sitting drop / pH: 7.5 Details: 0.05 M HEPES-CsOH pH 7.5, 0.2 M calcium chloride and 25% polyethylene glycol 4000 |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: BESSY  / Beamline: 14.1 / Wavelength: 0.917 Å / Beamline: 14.1 / Wavelength: 0.917 Å |
| Detector | Type: DECTRIS PILATUS 6M / Detector: PIXEL / Date: Nov 24, 2013 |
| Radiation | Monochromator: KMC-1 / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.917 Å / Relative weight: 1 |
| Reflection | Resolution: 1.45→40 Å / Num. obs: 87629 / % possible obs: 99.7 % / Redundancy: 6.8 % / CC1/2: 0.998 / Rmerge(I) obs: 0.121 / Rsym value: 0.112 / Net I/σ(I): 12 |
| Reflection shell | Resolution: 1.45→1.54 Å / Redundancy: 6.5 % / Rmerge(I) obs: 0.706 / Mean I/σ(I) obs: 2.5 / CC1/2: 0.813 / Rsym value: 0.649 / % possible all: 98.1 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 2B0P Resolution: 1.5→36.3 Å / SU ML: 0.12 / Cross valid method: FREE R-VALUE / σ(F): 1.36 / Phase error: 17.15 / Stereochemistry target values: ML Details: The N-terminus of the truncated protein (absent in the full length native LytM) is disordered and has been built in only tentatively. The ligand could be reliably modelled in the C-terminal ...Details: The N-terminus of the truncated protein (absent in the full length native LytM) is disordered and has been built in only tentatively. The ligand could be reliably modelled in the C-terminal and phosphinate part, its N-terminus is most likely disordered, since none of its conformations was clearly prevailing in all four molecules in the asymmetric unit. There are numerous close contacts between solvent molecules in the structure which might either result from their static disorder, or poorly ordered PEG molecules, or low occupancy metal ions all of which have been modelled tentatively in a few cases. Alternatively they may result from crystal defects (crystallites of different symmetries forming the macroscopically observed crystal). The identity of a few ions is unsure - for the technical reasons the unsure Cl- ions were kept as Cl ions whereas the unsure Na ions have been submitted as UNL atoms. For a few serine residues we observe difference density close to the side chain oxygen atom.
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.5→36.3 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
