 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-4wlj: High resolution crystal structure of human kynurenine aminotransf... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4wlj | ||||||
|---|---|---|---|---|---|---|---|
| Title | High resolution crystal structure of human kynurenine aminotransferase-I in complex with aminooxyacetate | ||||||
 Components Components | Kynurenine--oxoglutarate transaminase 1 | ||||||
 Keywords Keywords | TRANSFERASE / Lysine modification / Transaminase / Aminotransferase / Alpha beta protein Ligand / AOAA / Deoxy Aminopyridoxal Phosphate | ||||||
| Function / homology |  Function and homology information Function and homology informationglutamine-phenylpyruvate transaminase / glutamine-phenylpyruvate transaminase activity / L-kynurenine catabolic process / cysteine-S-conjugate beta-lyase / cysteine-S-conjugate beta-lyase activity / Phenylalanine metabolism / kynurenine-oxoglutarate transaminase / kynurenine-oxoglutarate transaminase activity / kynurenine metabolic process / Glutamate and glutamine metabolism ...glutamine-phenylpyruvate transaminase / glutamine-phenylpyruvate transaminase activity / L-kynurenine catabolic process / cysteine-S-conjugate beta-lyase / cysteine-S-conjugate beta-lyase activity / Phenylalanine metabolism / kynurenine-oxoglutarate transaminase / kynurenine-oxoglutarate transaminase activity / kynurenine metabolic process / Glutamate and glutamine metabolism / Tryptophan catabolism / biosynthetic process / response to bacterium / pyridoxal phosphate binding / protein homodimerization activity / mitochondrion / cytosol / cytoplasm Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 1.54 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 1.54 Å | ||||||
| Model details | The inhibitor aminooxyacetate forms an oxime with cofactor PLP that displaces the Schiff-base ...The inhibitor aminooxyacetate forms an oxime with cofactor PLP that displaces the Schiff-base linkage with the catalytic lysine247 | ||||||
 Authors Authors | Nadvi, N.A. / Salam, N.K. / Park, J. / Akladios, F.N. / Kapoor, V. / Collyer, C.A. / Gorrell, M.D. / Church, W.B. | ||||||
 Citation Citation | Journal: Protein Sci. / Year: 2017 Title: High resolution crystal structures of human kynurenine aminotransferase-I bound to PLP cofactor, and in complex with aminooxyacetate. Authors: Nadvi, N.A. / Salam, N.K. / Park, J. / Akladios, F.N. / Kapoor, V. / Collyer, C.A. / Gorrell, M.D. / Church, W.B. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4wlj.cif.gz | 321 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4wlj.ent.gz | 262.2 KB | Display | PDB format |
| PDBx/mmJSON format | 4wlj.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Summary document | 4wlj_validation.pdf.gz | 1.1 MB | Display | wwPDB validaton report |
|---|---|---|---|---|
| Full document | 4wlj_full_validation.pdf.gz | 1.1 MB | Display | |
| Data in XML | 4wlj_validation.xml.gz | 32.7 KB | Display | |
| Data in CIF | 4wlj_validation.cif.gz | 48.3 KB | Display | |
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/wl/4wljftp://data.pdbj.org/pub/pdb/validation_reports/wl/4wlj | HTTPS FTP |
-Related structure data
| Related structure data |  4wlhSC S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Details | biological unit is the same as asym. |
-Components
| #1: Protein | Mass: 47927.879 Da / Num. of mol.: 2 / Fragment: unp residues 87-392 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: CCBL1 / Plasmid: PBLUEBAC4.5 / Production host:   Spodoptera frugiperda (fall armyworm) Spodoptera frugiperda (fall armyworm)References: UniProt: Q16773, kynurenine-oxoglutarate transaminase, cysteine-S-conjugate beta-lyase, glutamine-phenylpyruvate transaminase #2: Chemical | 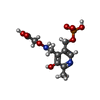  Mass: 322.208 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H15N2O8P Mass: 322.208 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H15N2O8P#3: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 392 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 392 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.19 Å3/Da / Density % sol: 43.74 % |
|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion, hanging drop / pH: 7.3 Details: 5 mM AOAA, 29% PEG 4000, 0.2 M sodium acetate, 0.1 M Tris |
-Data collection
| Diffraction | Mean temperature: 100 K | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: Australian Synchrotron  / Beamline: MX1 / Wavelength: 0.95369 Å / Beamline: MX1 / Wavelength: 0.95369 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Detector | Type: ADSC QUANTUM 210r / Detector: CCD / Date: Apr 2, 2011 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation | Monochromator: Silicon Double Crystal / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 0.95369 Å / Relative weight: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Resolution: 1.54→75.262 Å / Num. all: 108232 / Num. obs: 108232 / % possible obs: 89.8 % / Redundancy: 6.3 % / Biso Wilson estimate: 14.48 Å2 / Rpim(I) all: 0.033 / Rrim(I) all: 0.086 / Rsym value: 0.079 / Net I/av σ(I): 7.198 / Net I/σ(I): 13.2 / Num. measured all: 683702 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection shell | Diffraction-ID: 1 / Rejects: _
|
-Phasing
| Phasing | Method: molecular replacement |
|---|
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 4WLH Resolution: 1.54→26.94 Å / SU ML: 0.18 / Cross valid method: FREE R-VALUE / σ(F): 1.34 / Phase error: 22.47 / Stereochemistry target values: ML Details: The high resolution at which this structure was determined allowed the identification of residues with alternate conformations. Four regions in each chain were observed to be highly ...Details: The high resolution at which this structure was determined allowed the identification of residues with alternate conformations. Four regions in each chain were observed to be highly disordered with generally poorer election density: residues 15-29, 147-154, 355-359 and the C-terminus (419-422). Residues 147-154 were removed from the final model due to the absence of unambiguous election density. For residues in the other disordered regions electron density could be observed for most of the main chain atoms, but was less unambiguous for the side-chains. These residues are in the final model, with both the occupancy and temperature factors set to zero for most of these residues, due to the existence of a level of evidence for their location.
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 79.64 Å2 / Biso mean: 18.5506 Å2 / Biso min: 7.07 Å2 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 1.54→26.94 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION / Total num. of bins used: 30
|
