- PDB-4tz4: Crystal Structure of Human Cereblon in Complex with DDB1 and Lena... -
+
Open data
ID or keywords:
Loading...
-
Basic information
Entry
Database: PDB / ID: 4tz4
Title
Crystal Structure of Human Cereblon in Complex with DDB1 and Lenalidomide
Components
DNA damage-binding protein 1
Protein cereblon
Keywords
DNA Binding Protein/Ligase / DCAF / DNA Binding Protein-Ligase complex
Function / homology
Function and homology information
negative regulation of monoatomic ion transmembrane transport / positive regulation by virus of viral protein levels in host cell / spindle assembly involved in female meiosis / epigenetic programming in the zygotic pronuclei / UV-damage excision repair / biological process involved in interaction with symbiont / regulation of mitotic cell cycle phase transition / WD40-repeat domain binding / limb development / Cul4A-RING E3 ubiquitin ligase complex ...negative regulation of monoatomic ion transmembrane transport / positive regulation by virus of viral protein levels in host cell / spindle assembly involved in female meiosis / epigenetic programming in the zygotic pronuclei / UV-damage excision repair / biological process involved in interaction with symbiont / regulation of mitotic cell cycle phase transition / WD40-repeat domain binding / limb development / Cul4A-RING E3 ubiquitin ligase complex / Cul4-RING E3 ubiquitin ligase complex / negative regulation of reproductive process / negative regulation of developmental process / Cul4B-RING E3 ubiquitin ligase complex / ectopic germ cell programmed cell death / ubiquitin ligase complex scaffold activity / locomotory exploration behavior / viral release from host cell / cullin family protein binding / positive regulation of Wnt signaling pathway / negative regulation of protein-containing complex assembly / positive regulation of viral genome replication / positive regulation of gluconeogenesis / sperm end piece / sperm principal piece / nucleotide-excision repair / proteasomal protein catabolic process / positive regulation of protein-containing complex assembly / Recognition of DNA damage by PCNA-containing replication complex / regulation of circadian rhythm / Wnt signaling pathway / DNA Damage Recognition in GG-NER / Dual Incision in GG-NER / Transcription-Coupled Nucleotide Excision Repair (TC-NER) / Formation of TC-NER Pre-Incision Complex / Formation of Incision Complex in GG-NER / positive regulation of protein catabolic process / cellular response to UV / Dual incision in TC-NER / Gap-filling DNA repair synthesis and ligation in TC-NER / rhythmic process / sperm midpiece / site of double-strand break / Neddylation / Potential therapeutics for SARS / damaged DNA binding / ubiquitin-dependent protein catabolic process / proteasome-mediated ubiquitin-dependent protein catabolic process / transmembrane transporter binding / protein-macromolecule adaptor activity / chromosome, telomeric region / protein ubiquitination / DNA repair / apoptotic process / DNA damage response / nucleolus / negative regulation of apoptotic process / protein-containing complex binding / perinuclear region of cytoplasm / protein-containing complex / DNA binding / : / extracellular exosome / nucleoplasm / membrane / metal ion binding / nucleus / cytosol / cytoplasm Similarity search - Function
Type: MARMOSAIC 300 mm CCD / Detector: CCD / Date: Jan 15, 2014
Radiation
Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray
Radiation wavelength
Wavelength: 1 Å / Relative weight: 1
Reflection
Resolution: 3.01→50 Å / Num. obs: 37313 / % possible obs: 98.5 % / Redundancy: 6.1 % / Rmerge(I) obs: 0.181 / Χ2: 1.058 / Net I/av σ(I): 10.061 / Net I/σ(I): 5.9 / Num. measured all: 228575
Reflection shell
Diffraction-ID: 1 / Rejects: _
Resolution (Å)
Redundancy (%)
Rmerge(I) obs
Num. unique all
Χ2
% possible all
3.01-3.12
5.8
0.723
3184
1.02
85.8
3.12-3.24
6.3
0.584
3703
1.082
99.9
3.24-3.39
6.3
0.392
3731
1.08
100
3.39-3.57
6.3
0.294
3739
1.054
100
3.57-3.79
6.3
0.23
3768
1.045
100
3.79-4.08
6.2
0.187
3745
1.045
99.9
4.08-4.5
6.2
0.147
3795
1.034
100
4.5-5.15
6.1
0.121
3796
1.088
99.9
5.15-6.48
6.1
0.136
3835
1.087
99.9
6.48-50
5.6
0.082
4017
1.036
99.5
-
Processing
Software
Name
Version
Classification
REFMAC
5.6.0117
refinement
HKL-3000
datareduction
PDB_EXTRACT
3.14
dataextraction
Coot
modelbuilding
Refinement
Method to determine structure: MOLECULAR REPLACEMENT Starting model: DDB1 Resolution: 3.01→50 Å / Cor.coef. Fo:Fc: 0.918 / Cor.coef. Fo:Fc free: 0.873 / SU B: 38.571 / SU ML: 0.328 / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R Free: 0.465 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: HYDROGENS HAVE BEEN USED IF PRESENT IN THE INPUT U VALUES : RESIDUAL ONLY
Rfactor
Num. reflection
% reflection
Selection details
Rfree
0.2706
1857
5 %
RANDOM
Rwork
0.2015
35396
-
-
obs
0.2049
37253
99.4 %
-
Solvent computation
Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information Homo sapiens (human)
Homo sapiens (human) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads









 PDBj
PDBj



 Assembly
Assembly

 Spodoptera frugiperda (fall armyworm) / References: UniProt: Q16531
Spodoptera frugiperda (fall armyworm) / References: UniProt: Q16531
 Mass: 65.409 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Zn
Mass: 65.409 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Zn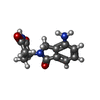
 Mass: 259.261 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C13H13N3O3
Mass: 259.261 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C13H13N3O3 Mass: 18.015 Da / Num. of mol.: 18 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 18 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation / Beamline: 08ID-1 / Wavelength: 1 Å
/ Beamline: 08ID-1 / Wavelength: 1 Å Processing
Processing