- PDB-4ci1: Structure of the DDB1-CRBN E3 ubiquitin ligase bound to thalidomide -
+
Open data
ID or keywords:
Loading...
-
Basic information
Entry
Database: PDB / ID: 4ci1
Title
Structure of the DDB1-CRBN E3 ubiquitin ligase bound to thalidomide
Components
DNA DAMAGE-BINDING PROTEIN 1
PROTEIN CEREBLON
Keywords
DNA BINDING PROTEIN/PROTEIN / DNA BINDING PROTEIN-PROTEIN COMPLEX / DNA BINDING PROTEIN-PROTEIN BINDING COMPLEX / UBIQUITIN / CONT
Function / homology
Function and homology information
positive regulation by virus of viral protein levels in host cell / spindle assembly involved in female meiosis / epigenetic programming in the zygotic pronuclei / UV-damage excision repair / limb development / biological process involved in interaction with symbiont / regulation of mitotic cell cycle phase transition / WD40-repeat domain binding / Cul4A-RING E3 ubiquitin ligase complex / Cul4-RING E3 ubiquitin ligase complex ...positive regulation by virus of viral protein levels in host cell / spindle assembly involved in female meiosis / epigenetic programming in the zygotic pronuclei / UV-damage excision repair / limb development / biological process involved in interaction with symbiont / regulation of mitotic cell cycle phase transition / WD40-repeat domain binding / Cul4A-RING E3 ubiquitin ligase complex / Cul4-RING E3 ubiquitin ligase complex / Cul4B-RING E3 ubiquitin ligase complex / ubiquitin ligase complex scaffold activity / negative regulation of reproductive process / negative regulation of developmental process / viral release from host cell / cullin family protein binding / ectopic germ cell programmed cell death / positive regulation of Wnt signaling pathway / positive regulation of viral genome replication / positive regulation of gluconeogenesis / proteasomal protein catabolic process / sperm end piece / nucleotide-excision repair / sperm principal piece / regulation of circadian rhythm / Recognition of DNA damage by PCNA-containing replication complex / DNA Damage Recognition in GG-NER / Wnt signaling pathway / Dual Incision in GG-NER / Transcription-Coupled Nucleotide Excision Repair (TC-NER) / Formation of TC-NER Pre-Incision Complex / Formation of Incision Complex in GG-NER / positive regulation of protein catabolic process / cellular response to UV / Dual incision in TC-NER / Gap-filling DNA repair synthesis and ligation in TC-NER / rhythmic process / sperm midpiece / site of double-strand break / Neddylation / ubiquitin-dependent protein catabolic process / damaged DNA binding / proteasome-mediated ubiquitin-dependent protein catabolic process / protein-macromolecule adaptor activity / chromosome, telomeric region / protein ubiquitination / DNA repair / apoptotic process / DNA damage response / negative regulation of apoptotic process / nucleolus / protein-containing complex binding / protein-containing complex / : / DNA binding / extracellular exosome / nucleoplasm / metal ion binding / nucleus / cytoplasm Similarity search - Function
DNADAMAGE-BINDINGPROTEIN1 / DDB P127 SUBUNIT / DNA DAMAGE-BINDING PROTEIN A / DDBA / DAMAGE-SPECIFIC DNA-BINDING PROTEIN 1 / ...DDB P127 SUBUNIT / DNA DAMAGE-BINDING PROTEIN A / DDBA / DAMAGE-SPECIFIC DNA-BINDING PROTEIN 1 / HBV X-ASSOCIATED PROTEIN 1 / XAP-1 / UV-DAMAGED DNA-BINDING FACTOR / UV-DAMAGED DNA-BINDING PROTEIN 1 / UV-DDB 1 / XPE-BINDING FACTOR / XPE-BF / XERODERMA PIGMENTOSUM GROUP E-COMPLEMENTING PROTEIN / XPCE
Mass: 129493.852 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) HOMO SAPIENS (human) / Plasmid: PFASTBAC DUAL / Cell line (production host): High Five / Production host: TRICHOPLUSIA NI (cabbage looper) / References: UniProt: Q16531
#2: Protein
PROTEINCEREBLON
Mass: 54207.148 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) GALLUS GALLUS (chicken) / Plasmid: PFASTBAC DUAL / Cell line (production host): High Five / Production host: TRICHOPLUSIA NI (cabbage looper) / References: UniProt: P0CF65
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information HOMO SAPIENS (human)
HOMO SAPIENS (human)
 X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads









 PDBj
PDBj



 Assembly
Assembly
 TRICHOPLUSIA NI (cabbage looper) / References: UniProt: Q16531
TRICHOPLUSIA NI (cabbage looper) / References: UniProt: Q16531
 Mass: 65.409 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Zn
Mass: 65.409 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Zn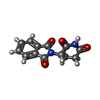
 Mass: 258.229 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C13H10N2O4 / Comment: medication*YM
Mass: 258.229 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C13H10N2O4 / Comment: medication*YM Mass: 18.015 Da / Num. of mol.: 35 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 35 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation / Beamline: X10SA / Wavelength: 0.99999
/ Beamline: X10SA / Wavelength: 0.99999  Processing
Processing