解像度: 1.75→24.07 Å / Cor.coef. Fo:Fc: 0.97 / Cor.coef. Fo:Fc free: 0.949 / WRfactor Rfree: 0.2294 / WRfactor Rwork: 0.1496 / Occupancy max: 1 / Occupancy min: 0.3 / FOM work R set: 0.7967 / SU B: 3.803 / SU ML: 0.115 / SU R Cruickshank DPI: 0.1368 / SU Rfree: 0.1482 / 交差検証法: THROUGHOUT / σ(F): 0 / ESU R: 0.137 / ESU R Free: 0.148 / 立体化学のターゲット値: MAXIMUM LIKELIHOOD 詳細: Model is based on initial rigid body refinement of protein coordinates from PDB entry 1MHN. SMILES(CN1C2=C(CCC2)C(=N)C2=C1CCC2) represents the uncharged form of the ligand. Geometry ...詳細: Model is based on initial rigid body refinement of protein coordinates from PDB entry 1MHN. SMILES(CN1C2=C(CCC2)C(=N)C2=C1CCC2) represents the uncharged form of the ligand. Geometry restraints for the inhibitor were prepared on the GRADE server with SMILES(C[n+]1c2CCCc2c(N)c2CCCc12). We note that mogul (CSD) inferred the type of the C1-C2 bond in refined ligand coordinates as double, which would be inconsistent with any traditional hybridization state of C1. COOT and MOLPROBITY were also used during refinement.
Rfactor
反射数
%反射
Selection details
Rfree
0.2417
508
10.3 %
RANDOM
Rwork
0.1646
-
-
-
obs
0.1727
4942
99.64 %
-
溶媒の処理
イオンプローブ半径: 0.8 Å / 減衰半径: 0.8 Å / VDWプローブ半径: 1.2 Å / 溶媒モデル: MASK
 ムービー
ムービー コントローラー
コントローラー


 データを開く
データを開く
 基本情報
基本情報 要素
要素 キーワード
キーワード 機能・相同性情報
機能・相同性情報 Homo sapiens (ヒト)
Homo sapiens (ヒト) X線回折 /
X線回折 /  データ登録者
データ登録者 引用
引用 構造の表示
構造の表示 ダウンロードとリンク
ダウンロードとリンク その他のダウンロード
その他のダウンロード









 PDBj
PDBj
 集合体
集合体


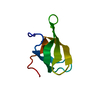
 分子数: 3 / 由来タイプ: 合成
分子数: 3 / 由来タイプ: 合成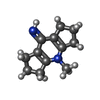
 分子量: 188.269 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C12H16N2
分子量: 188.269 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C12H16N2 分子量: 18.015 Da / 分子数: 26 / 由来タイプ: 天然 / 式: H2O
分子量: 18.015 Da / 分子数: 26 / 由来タイプ: 天然 / 式: H2O 試料調製
試料調製 解析
解析