 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-4qq6: Crystal Structure of tudor domain of SMN1 in complex with a small... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4qq6 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal Structure of tudor domain of SMN1 in complex with a small organic molecule | ||||||
 Components Components | Survival motor neuron protein | ||||||
 Keywords Keywords | RNA BINDING PROTEIN / structural genomics / Structural Genomics Consortium / SGC | ||||||
| Function / homology |  Function and homology information Function and homology informationGemini of Cajal bodies / SMN complex / SMN-Sm protein complex / spliceosomal complex assembly / spliceosomal snRNP assembly / Cajal body / DNA-templated transcription termination / Z disc / cytoplasmic ribonucleoprotein granule / nervous system development ...Gemini of Cajal bodies / SMN complex / SMN-Sm protein complex / spliceosomal complex assembly / spliceosomal snRNP assembly / Cajal body / DNA-templated transcription termination / Z disc / cytoplasmic ribonucleoprotein granule / nervous system development / snRNP Assembly / SARS-CoV-2 modulates host translation machinery / perikaryon / neuron projection / nuclear body / axon / RNA binding / nucleoplasm / identical protein binding / nucleus / cytoplasm / cytosol Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 1.75 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 1.75 Å | ||||||
 Authors Authors | Liu, Y. / Tempel, W. / Iqbal, A. / Walker, J.R. / Bountra, C. / Arrowsmith, C.H. / Edwards, A.M. / Brown, P.J. / Min, J. / Structural Genomics Consortium (SGC) | ||||||
 Citation Citation | Journal: Nat Commun / Year: 2022 Title: A small molecule antagonist of SMN disrupts the interaction between SMN and RNAP II. Authors: Liu, Y. / Iqbal, A. / Li, W. / Ni, Z. / Wang, Y. / Ramprasad, J. / Abraham, K.J. / Zhang, M. / Zhao, D.Y. / Qin, S. / Loppnau, P. / Jiang, H. / Guo, X. / Brown, P.J. / Zhen, X. / Xu, G. / ...Authors: Liu, Y. / Iqbal, A. / Li, W. / Ni, Z. / Wang, Y. / Ramprasad, J. / Abraham, K.J. / Zhang, M. / Zhao, D.Y. / Qin, S. / Loppnau, P. / Jiang, H. / Guo, X. / Brown, P.J. / Zhen, X. / Xu, G. / Mekhail, K. / Ji, X. / Bedford, M.T. / Greenblatt, J.F. / Min, J. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4qq6.cif.gz | 26.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4qq6.ent.gz | 15.7 KB | Display | PDB format |
| PDBx/mmJSON format | 4qq6.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/qq/4qq6ftp://data.pdbj.org/pub/pdb/validation_reports/qq/4qq6 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  4qqdC  6v9tC  7w2pC  7w30C  1mhnS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |







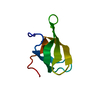
-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Details | THE BIOLOGICAL UNIT IS UNKNOWN |
-Components
| #1: Protein | Mass: 7409.304 Da / Num. of mol.: 1 / Fragment: unp residues 82-147 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: SMN1, SMN, SMNT, SMN2, SMNC / Plasmid: pET28-MHL / Production host:  | ||||
|---|---|---|---|---|---|
| #2: Chemical |   Num. of mol.: 3 / Source method: obtained synthetically Num. of mol.: 3 / Source method: obtained synthetically#3: Chemical | ChemComp-36X / | 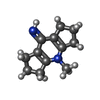  Mass: 188.269 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C12H16N2 Mass: 188.269 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C12H16N2#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 26 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 26 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 1.7 Å3/Da / Density % sol: 27.53 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, sitting drop / pH: 5.6 Details: 2M ammonium sulfate, 0.2M potassium/sodium tartrate, 0.1M sodium citrate, pH 5.6, VAPOR DIFFUSION, SITTING DROP, temperature 293K |
-Data collection
| Diffraction | Mean temperature: 100 K | |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU FR-E / Wavelength: 1.5418 Å | |||||||||||||||||||||
| Detector | Type: RIGAKU SATURN A200 / Detector: CCD / Date: Apr 15, 2014 | |||||||||||||||||||||
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | |||||||||||||||||||||
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 | |||||||||||||||||||||
| Reflection | Resolution: 1.75→37.61 Å / Num. obs: 5006 / % possible obs: 100 % / Redundancy: 10.5 % / Rmerge(I) obs: 0.078 / Net I/σ(I): 19.8 | |||||||||||||||||||||
| Reflection shell | Diffraction-ID: 1
|
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: pdb entry 1MHN Resolution: 1.75→24.07 Å / Cor.coef. Fo:Fc: 0.97 / Cor.coef. Fo:Fc free: 0.949 / WRfactor Rfree: 0.2294 / WRfactor Rwork: 0.1496 / Occupancy max: 1 / Occupancy min: 0.3 / FOM work R set: 0.7967 / SU B: 3.803 / SU ML: 0.115 / SU R Cruickshank DPI: 0.1368 / SU Rfree: 0.1482 / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R: 0.137 / ESU R Free: 0.148 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: Model is based on initial rigid body refinement of protein coordinates from PDB entry 1MHN. SMILES(CN1C2=C(CCC2)C(=N)C2=C1CCC2) represents the uncharged form of the ligand. Geometry ...Details: Model is based on initial rigid body refinement of protein coordinates from PDB entry 1MHN. SMILES(CN1C2=C(CCC2)C(=N)C2=C1CCC2) represents the uncharged form of the ligand. Geometry restraints for the inhibitor were prepared on the GRADE server with SMILES(C[n+]1c2CCCc2c(N)c2CCCc12). We note that mogul (CSD) inferred the type of the C1-C2 bond in refined ligand coordinates as double, which would be inconsistent with any traditional hybridization state of C1. COOT and MOLPROBITY were also used during refinement.
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 72.38 Å2 / Biso mean: 26.6793 Å2 / Biso min: 14.92 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.75→24.07 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.75→1.795 Å / Total num. of bins used: 20
|
