| Entry | Database: PDB / ID: 4q93
|
|---|
| Title | Crystal structure of resveratrol bound human tyrosyl tRNA synthetase |
|---|
 Components Components | Tyrosine--tRNA ligase, cytoplasmic |
|---|
 Keywords Keywords | LIGASE / Resveratrol / Aminoacyl tRNA synthetases / Amino acid activation / active site |
|---|
| Function / homology |  Function and homology information Function and homology information
interleukin-8 receptor binding / tyrosyl-tRNA aminoacylation / tyrosine-tRNA ligase / tyrosine-tRNA ligase activity / Cytosolic tRNA aminoacylation / response to starvation / small molecule binding / tRNA binding / nuclear body / apoptotic process ...interleukin-8 receptor binding / tyrosyl-tRNA aminoacylation / tyrosine-tRNA ligase / tyrosine-tRNA ligase activity / Cytosolic tRNA aminoacylation / response to starvation / small molecule binding / tRNA binding / nuclear body / apoptotic process / : / RNA binding / ATP binding / nucleus / cytoplasm / cytosolSimilarity search - Function : / Tyrosine-tRNA ligase / Tyrosyl-Transfer RNA Synthetase / Tyrosyl-Transfer RNA Synthetase / tRNA-binding domain / Putative tRNA binding domain / tRNA-binding domain profile. / Aminoacyl-tRNA synthetase, class Ic / tRNA synthetases class I (W and Y) / HUPs ...: / Tyrosine-tRNA ligase / Tyrosyl-Transfer RNA Synthetase / Tyrosyl-Transfer RNA Synthetase / tRNA-binding domain / Putative tRNA binding domain / tRNA-binding domain profile. / Aminoacyl-tRNA synthetase, class Ic / tRNA synthetases class I (W and Y) / HUPs / Rossmann-like alpha/beta/alpha sandwich fold / Nucleic acid-binding, OB-fold / Rossmann fold / Orthogonal Bundle / 3-Layer(aba) Sandwich / Mainly Alpha / Alpha BetaSimilarity search - Domain/homology |
|---|
| Biological species |  Homo sapiens (human) Homo sapiens (human) |
|---|
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.1 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.1 Å |
|---|
 Authors Authors | Mathew, S. / Schimmel, P. |
|---|
 Citation Citation | Journal: Nature / Year: 2014
Title: A human tRNA synthetase is a potent PARP1-activating effector target for resveratrol.
Authors: Sajish, M. / Schimmel, P. |
|---|
| History | | Deposition | Apr 28, 2014 | Deposition site: RCSB / Processing site: RCSB |
|---|
| Revision 1.0 | Dec 17, 2014 | Provider: repository / Type: Initial release |
|---|
| Revision 1.1 | Dec 24, 2014 | Group: Structure summary |
|---|
| Revision 1.2 | Jan 14, 2015 | Group: Database references |
|---|
| Revision 1.3 | Mar 25, 2015 | Group: Database references |
|---|
| Revision 1.4 | Jul 17, 2019 | Group: Data collection / Refinement description / Category: software
Item: _software.classification / _software.name / _software.version |
|---|
| Revision 1.5 | Sep 20, 2023 | Group: Data collection / Database references ...Data collection / Database references / Derived calculations / Refinement description
Category: chem_comp_atom / chem_comp_bond ...chem_comp_atom / chem_comp_bond / database_2 / pdbx_initial_refinement_model / struct_ref_seq_dif / struct_site
Item: _database_2.pdbx_DOI / _database_2.pdbx_database_accession ..._database_2.pdbx_DOI / _database_2.pdbx_database_accession / _struct_ref_seq_dif.details / _struct_site.pdbx_auth_asym_id / _struct_site.pdbx_auth_comp_id / _struct_site.pdbx_auth_seq_id |
|---|
|
|---|
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads

















 PDBj
PDBj

 Assembly
Assembly


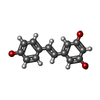





 Mass: 228.243 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C14H12O3
Mass: 228.243 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C14H12O3 Mass: 96.063 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: SO4
Mass: 96.063 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: SO4 Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl
Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl Mass: 92.094 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C3H8O3
Mass: 92.094 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 94.971 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: PO4
Mass: 94.971 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: PO4 Sample preparation
Sample preparation Processing
Processing