 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-4m7y: Staphylococcus aureus Type II pantothenate kinase in complex with... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4m7y | ||||||
|---|---|---|---|---|---|---|---|
| Title | Staphylococcus aureus Type II pantothenate kinase in complex with a pantothenate analog | ||||||
 Components Components | Type II pantothenate kinase | ||||||
 Keywords Keywords | TRANSFERASE/TRANSFERASE INHIBITOR / pantothenate kinase / inhibitor / antibacterial / Structural Genomics / Structural Genomics Consortium / SGC / TRANSFERASE-TRANSFERASE INHIBITOR complex | ||||||
| Function / homology |  Function and homology information Function and homology informationpantothenate kinase / pantothenate kinase activity / coenzyme A biosynthetic process / ATP binding / cytosol Similarity search - Function | ||||||
| Biological species |   Staphylococcus aureus subsp. aureus (bacteria) Staphylococcus aureus subsp. aureus (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 1.8 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 1.8 Å | ||||||
 Authors Authors | Mottaghi, K. / Hong, B. / Tempel, W. / Park, H. / Structural Genomics Consortium (SGC) | ||||||
 Citation Citation | Journal: ACS Infect Dis / Year: 2016 Title: Discovery of Potent Pantothenamide Inhibitors of Staphylococcus aureus Pantothenate Kinase through a Minimal SAR Study: Inhibition Is Due to Trapping of the Product. Authors: Hughes, S.J. / Barnard, L. / Mottaghi, K. / Tempel, W. / Antoshchenko, T. / Hong, B.S. / Allali-Hassani, A. / Smil, D. / Vedadi, M. / Strauss, E. / Park, H.W. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4m7y.cif.gz | 227.6 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4m7y.ent.gz | 180.8 KB | Display | PDB format |
| PDBx/mmJSON format | 4m7y.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/m7/4m7yftp://data.pdbj.org/pub/pdb/validation_reports/m7/4m7y | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  4m7xSC  5elzC  5jicC S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Components on special symmetry positions |
| ||||||||
| Details | AS PER THE AUTHORS THE BIOLOGICAL ASSEMBLY IS UNKNOWN |
-Components
-Protein , 1 types, 2 molecules AB
| #1: Protein | Mass: 29955.818 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Staphylococcus aureus subsp. aureus (bacteria)Strain: MW2 / Gene: coaW, MW2054 / Production host: |
|---|
-Non-polymers , 5 types, 226 molecules 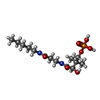




| #2: Chemical |  Mass: 368.363 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C14H29N2O7P Mass: 368.363 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C14H29N2O7P#3: Chemical |  Mass: 427.201 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H15N5O10P2 / Comment: ADP, energy-carrying molecule*YM Mass: 427.201 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H15N5O10P2 / Comment: ADP, energy-carrying molecule*YM#4: Chemical | ChemComp-PO4 /  Mass: 94.971 Da / Num. of mol.: 7 / Source method: obtained synthetically / Formula: PO4 Mass: 94.971 Da / Num. of mol.: 7 / Source method: obtained synthetically / Formula: PO4#5: Chemical | ChemComp-UNX /  Num. of mol.: 22 / Source method: obtained synthetically Num. of mol.: 22 / Source method: obtained synthetically#6: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 193 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.3 Å3/Da / Density % sol: 46.3 % |
|---|---|
| Crystal grow | Method: vapor diffusion / pH: 7 Details: 0.01 M inhibitor, 0.01 M magnesium chloride, 0.01 M ATP were added to the protein sample. Crystal condition: 2.5 M ammonium sulfate, 0.1 M BTP, 1.1 % w/v Cyclohexylethanoyl-N- ...Details: 0.01 M inhibitor, 0.01 M magnesium chloride, 0.01 M ATP were added to the protein sample. Crystal condition: 2.5 M ammonium sulfate, 0.1 M BTP, 1.1 % w/v Cyclohexylethanoyl-N-hydroxyethylglucamide., pH 7.0, vapor diffusion |
-Data collection
| Diffraction | Mean temperature: 100 K | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU FR-E / Wavelength: 1.5418 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Detector | Type: RIGAKU RAXIS / Detector: IMAGE PLATE / Date: Mar 24, 2011 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Resolution: 1.8→30 Å / Num. obs: 48878 / % possible obs: 99.4 % / Observed criterion σ(I): -3 / Biso Wilson estimate: 33.94 Å2 / Rmerge(I) obs: 0.079 / Net I/σ(I): 14.87 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection shell | Diffraction-ID: 1
|
-Phasing
| Phasing | Method: molecular replacement |
|---|
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB entry 4m7x Resolution: 1.8→26.532 Å / Cor.coef. Fo:Fc: 0.967 / Cor.coef. Fo:Fc free: 0.951 / WRfactor Rfree: 0.208 / WRfactor Rwork: 0.167 / Occupancy max: 1 / Occupancy min: 0.3 / SU B: 6.661 / SU ML: 0.099 / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R: 0.126 / ESU R Free: 0.124 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS U VALUES: WITH TLS ADDED. INHIBITOR RESTRAINTS WERE BASED ON PRODRG CALCULATIONS. COOT AND THE MOLPROBITY SERVER WERE ALSO USED DURING ...Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS U VALUES: WITH TLS ADDED. INHIBITOR RESTRAINTS WERE BASED ON PRODRG CALCULATIONS. COOT AND THE MOLPROBITY SERVER WERE ALSO USED DURING REFINEMENT. SOME DUMMY ATOMS AND PHOSPHATE IONS B303, B304, A303, B305 OF THE MODEL REPRESENT ELECTRON DENSITY THAT APPEARS TO ARISE FROM A LARGER, UNIDENTIFIED MOLECULE.
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK BULK SOLVENT | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 83.01 Å2 / Biso mean: 31.734 Å2 / Biso min: 15.15 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.8→26.532 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION / Total num. of bins used: 20
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
